Per segnalare un effetto indesiderato o un reclamo su un prodotto Lilly, si prega di non utilizzare questo sito web. La segnalazione delle reazioni avverse è estremamente importante. Gli operatori sanitari e i pazienti possono riportare una reazione avversa o un reclamo di un prodotto sul sito di AIFA.
▼Jaypirca® (pirtobrutinib)
Medicinale sottoposto a monitoraggio addizionale. Ciò permetterà la rapida identificazione di nuove informazioni sulla sicurezza. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta. Vedere paragrafo 4.8 per informazioni sulle modalità di segnalazione delle reazioni avverse.Jaypirca 50 mg compresse rivestite con film
Jaypirca 100 mg compresse rivestite con film
Jaypirca 50 mg compresse rivestite con film
Ogni compressa rivestita con film contiene 50 mg di pirtobrutinib.
Eccipienti con effetti noti
Ogni compressa rivestita con film contiene 38 mg di lattosio (come monoidrato).
Jaypirca 100 mg compresse rivestite con film
Ogni compressa rivestita con film contiene 100 mg di pirtobrutinib.
Eccipienti con effetti noti
Ogni compressa rivestita con film contiene 77 mg di lattosio (come monoidrato).
Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
Compressa rivestita con film (compressa).
Jaypirca 50 mg compresse rivestite con film
Compressa blu, di 9 x 9 mm, a forma di triangolo con bordi arcuati, con impresso "Lilly 50" su un lato e "6902" sull'altro.
Jaypirca 100 mg compresse rivestite con film
Compressa blu, di 10 mm, rotonda, con inciso “Lilly 100” su un lato e “7026” sull'altro.
4.1 Indicazioni terapeutiche
Jaypirca in monoterapia è indicato per il trattamento di pazienti adulti affetti da linfoma a cellule mantellari (mantle cell lymphoma, MCL) recidivante o refrattario che sono stati precedentemente trattati con un inibitore della tirosin chinasi di Bruton (Bruton’s tyrosine kinase, BTK).
Jaypirca in monoterapia è indicato per il trattamento di pazienti adulti affetti da leucemia linfatica cronica (chronic lymphocytic leukaemia, CLL) recidivante o refrattaria che sono stati precedentemente trattati con un inibitore di BTK.
4.2 Posologia e modo di somministrazione
La terapia con Jaypirca deve essere iniziata e supervisionata da medici esperti nell'uso di terapie antitumorali.
Posologia
La dose raccomandata è di 200 mg di pirtobrutinib una volta al giorno (QD).
La somministrazione di Jaypirca deve essere interrotta fino a risoluzione al grado 1 o al basale quando il paziente manifesta uno dei seguenti eventi:
• neutropenia di grado 3 con febbre e/o infezione
• neutropenia di grado 4 di durata ≥ 7 giorni
• trombocitopenia di grado 3 con sanguinamento
• trombocitopenia di grado 4
• tossicità non ematologica di grado 3 o 4
La linfocitosi asintomatica non è considerata una reazione avversa e i pazienti che manifestano questo evento devono continuare ad assumere Jaypirca.
Negli studi clinici, gli eventi avversi in un numero limitato di pazienti sono stati gestiti mediante la riduzione della dose (vedere paragrafo 5.1).
Il trattamento deve essere continuato fino a progressione di malattia o a tossicità inaccettabile.
Dose saltata
Se sono trascorse più di 12 ore da quando è stata saltata una dose, il paziente deve essere istruito a prendere la dose successiva all'orario programmato; non deve essere assunta una dose aggiuntiva. In caso di vomito, il paziente non deve assumere una dose aggiuntiva ma continuare con la dose successiva programmata.
Popolazioni speciali
Anziani
Non è richiesto alcun adeguamento della dose in base all'età (vedere paragrafo 5.2).
Compromissione renale
Non è richiesto alcun adeguamento della dose per i pazienti con compromissione renale lieve, moderata o severa. Non ci sono dati nei pazienti in dialisi (vedere paragrafo 5.2).
Compromissione epatica
Non è richiesto alcun adeguamento della dose per i pazienti con compromissione epatica lieve, moderata o severa (vedere paragrafo 5.2).
Popolazione pediatrica
La sicurezza e l’efficacia di Jaypirca nei bambini e negli adolescenti di età inferiore a 18 anni non sono state stabilite. Non ci sono dati disponibili.
Modo di somministrazione
Jaypirca è per uso orale.
La compressa deve essere deglutita intera con un bicchiere d’acqua per garantire un rendimento costante (i pazienti non devono masticare, frantumare o dividere le compresse prima di deglutirle) e può essere assunta con o senza cibo. I pazienti devono assumere la dose all'incirca alla stessa ora ogni giorno.
4.3 Controindicazioni
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
4.4 Avvertenze speciali e precauzioni d’impiego
Infezioni
Nei pazienti trattati con Jaypirca si sono verificate infezioni gravi, inclusi casi fatali. Le infezioni di grado 3 o superiore più frequentemente riportate sono state infezione polmonare, polmonite da COVID-19, COVID-19 e sepsi. La terapia antibiotica profilattica deve essere presa in considerazione nei pazienti a maggior rischio di infezioni opportunistiche. Sulla base del grado dell’infezione e se si
verifica con neutropenia, può essere necessaria l’interruzione della dose (vedere paragrafo 4.2).
Emorragia
Eventi di sanguinamento, inclusi casi fatali, si sono verificati nei pazienti trattati con Jaypirca, con e senza trombocitopenia. Sono stati osservati eventi di anguinamento maggiori, di grado 3 o superiore,
inclusi sanguinamento gastrointestinale ed emorragia intracranica. I pazienti devono essere monitorati per segni e sintomi di sanguinamento. I pazienti che assumono anticoagulanti o agenti antiaggreganti piastrinici possono essere a maggior rischio di emorragia. I rischi e i benefici della terapia anticoagulante o anti-piastrinica devono essere considerati quando somministrata insieme a Jaypirca e deve essere preso in considerazione un monitoraggio aggiuntivo per i segni di sanguinamento. L'uso di Jaypirca non è stato studiato con warfarin o altri antagonisti della vitamina K. Può essere necessaria l’interruzione della dose per eventi emorragici di grado 3 o 4 (vedere paragrafo 4.2).
Il rischio-beneficio della sospensione di Jaypirca per 3-5 giorni prima e dopo un intervento chirurgico deve essere considerato a seconda del tipo di intervento chirurgico e del rischio di sanguinamento.
Citopenie
Nei pazienti trattati con Jaypirca si sono verificate citopenie di grado 3 o 4, incluse neutropenia, anemia e trombocitopenia. L’esame emocromocitometrico deve essere monitorato durante il trattamento come clinicamente indicato. Può essere necessaria l’interruzione della dose in base al grado della citopenia (vedere paragrafo 4.2).
Fibrillazione atriale/flutter atriale
Fibrillazione atriale e flutter atriale sono stati osservati nei pazienti trattati con Jaypirca, in particolare nei pazienti con una storia di fibrillazione atriale e/o comorbidità cardiovascolari multiple. Devono essere monitorati i segni e i sintomi della fibrillazione atriale e del flutter atriale; richiedere un elettrocardiogramma come clinicamente indicato. Può essere necessaria l’interruzione della dose in
base al grado della fibrillazione atriale/flutter atriale (vedere paragrafo 4.2).
Seconde malignità primitive
Nei pazienti trattati con Jaypirca si sono comunemente verificate seconde malignità primitive, delle quali i tipi più frequenti sono i tumori cutanei non-melanoma. I pazienti devono essere monitorati per la comparsa di tumori della pelle e deve essere consigliata la protezione all'esposizione al sole.
Sindrome da lisi tumorale
La sindrome da lisi tumorale (Tumour lysis syndrome, TLS) è stata segnalata raramente durante la terapia con Jaypirca. I pazienti ad alto rischio di TLS sono quelli con elevato carico tumorale prima del trattamento. I pazienti devono essere valutati per il possibile rischio di TLS e attentamente
monitorati come clinicamente indicato.
Contraccezione nelle donne in età fertile e negli uomini
In base ai risultati negli animali e alla genotossicità di pirtobrutinib (vedere paragrafo 5.3), pirtobrutinib può causare danni al feto se somministrato a donne in gravidanza. Le donne in età fertile devono utilizzare un metodo contraccettivo efficace durante il trattamento e per 5 settimane dopo l'ultima dose di Jaypirca. Si consiglia agli uomini di utilizzare un metodo contraccettivo efficace e di
non procreare durante il trattamento e per 3 mesi dopo l'ultima dose di Jaypirca (vedere paragrafo 4.6).
Lattosio
I pazienti affetti da rari problemi ereditari di intolleranza al galattosio, da deficit totale di lattasi o da malassorbimento di glucosio-galattosio non devono assumere questo medicinale.
Sodio
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per dose giornaliera di 200 mg, cioè
essenzialmente ‘senza sodio’.
4.5 Interazioni con altri medicinali ed altre forme d’interazione
Pirtobrutinib è metabolizzato principalmente da CYP3A4, UGT1A8 e UGT1A9.
Effetti di altri medicinali sulla farmacocinetica di pirtobrutinib
Inibitori di CYP3A
In uno studio clinico, itraconazolo, un potente inibitore di CYP3A4, ha aumentato l'AUC di pirtobrutinib del 48% e non ha modificato la Cmax di pirtobrutinib. Questo aumento dell'esposizione a pirtobrutinib non è clinicamente significativo. Pertanto, non è necessario alcun adeguamento della dose di Jaypirca con gli inibitori di CYP3A.
Induttori di CYP3A
In uno studio clinico, rifampicina, un potente induttore di CYP3A, ha ridotto l'AUC e la Cmax di pirtobrutinib rispettivamente del 71 % e del 42 %. Sebbene non si preveda che questa riduzione dell'esposizione a pirtobrutinib sia clinicamente significativa, se possibile evitare forti induttori di CYP3A (ad es. rifampicina, carbamazepina, fenitoina).
Somministrazione concomitante con inibitori di pompa protonica
Non sono state osservate differenze clinicamente significative nella farmacocinetica di pirtobrutinib
quando somministrato in concomitanza con omeprazolo, un inibitore di pompa protonica.
Effetti di pirtobrutinib sulla farmacocinetica di altri medicinali (aumento della concentrazione
plasmatica)
Substrati di CYP2C8
Pirtobrutinib è un inibitore moderato di CYP2C8. Pirtobrutinib ha aumentato l'AUC e la Cmax di repaglinide (un substrato di CYP2C8) rispettivamente del 130 % e del 98 %. Pertanto, poiché pirtobrutinib può aumentare le concentrazioni plasmatiche dei substrati di CYP2C8, si consiglia cautela nella co-somministrazione con substrati di CYP2C8 (ad es. repaglinide, dasabuvir, selexipag,
rosiglitazone, pioglitazone e montelukast).
Substrati di BCRP
Pirtobrutinib è un inibitore moderato della proteina di resistenza del cancro della mammella (Breast Cancer Resistance Protein, BCRP). Pirtobrutinib ha aumentato l'AUC e la Cmax di rosuvastatina (un substrato di BCRP) rispettivamente del 140 % e del 146 %. Pertanto, poiché pirtobrutinib può aumentare le concentrazioni plasmatiche dei substrati di BCRP, si consiglia cautela nella co-somministrazione di substrati di BCRP (ad es. rosuvastatina). Se la somministrazione concomitante con substrati di BCRP con indice terapeutico ristretto (ad es. metotrexato ad alto dosaggio, mitoxantrone) non può essere evitata, deve essere preso in considerazione uno stretto monitoraggio clinico.
Substrati di P-gp
Pirtobrutinib è un debole inibitore della glicoproteina P (P-gp). Pirtobrutinib ha aumentato l'AUC e la Cmax della digossina (un substrato della P-gp) rispettivamente del 35 % e del 55 %. Pertanto, pirtobrutinib può aumentare le concentrazioni plasmatiche dei substrati di P-gp. Se la co-somministrazione con substrati di P-gp con indice Terapeutico ristretto (ad es. dabigatran etexilato e
digossina) non può essere evitata, deve essere preso in considerazione uno stretto monitoraggio clinico.
Substrati di CYP2C19
Pirtobrutinib è un inibitore debole di CYP2C19. Pirtobrutinib ha aumentato l'AUC e la Cmax di
omeprazolo (un substrato di CYP2C19) rispettivamente del 56 % e del 49 %. Pertanto, pirtobrutinib può aumentare le concentrazioni plasmatiche dei substrati di CYP2C19. Se la co-somministrazione con substrati di CYP2C19 con indice terapeutico ristretto (ad es. fenobarbital e mefenitoina) non può essere evitata, deve essere preso in considerazione uno stretto monitoraggio clinico.
Substrati di CYP3A
Pirtobrutinib è un inibitore debole di CYP3A. Pirtobrutinib ha aumentato l'AUC e la Cmax di midazolam somministrato per via orale (substrato sensibile di CYP3A) rispettivamente del 70 % e del 58 %. Pirtobrutinib non ha avuto un effetto clinicamente significativo sull'esposizione al midazolam
somministrato per via endovenosa. Pertanto, pirtobrutinib può aumentare le concentrazioni plasmatiche dei substrati di CYP3A. Se la co-somministrazione con substrati di CYP3A con indice terapeutico ristretto (ad es. alfentanil, midazolam, tacrolimus) non può essere evitata, deve essere
preso in considerazione uno stretto monitoraggio clinico.
4.6 Fertilità, gravidanza e allattamento
Donne in età fertile/contraccezione negli uomini e nelle donne
In base ai risultati negli animali e alla genotossicità di pirtobrutinib (vedere paragrafo 5.3), pirtobrutinib può causare danni al feto se somministrato a donne in gravidanza. Le donne in età fertile devono utilizzare un metodo contraccettivo efficace durante il trattamento e fino a 5 settimane dopo l'ultima dose di Jaypirca. Si consiglia agli uomini di utilizzare un metodo contraccettivo efficace e di non procreare durante il trattamento e per 3 mesi dopo l'ultima dose di Jaypirca (vedere paragrafo 4.4).
Gravidanza
Non esistono dati relativi all'uso di Jaypirca in donne in gravidanza. Gli studi sugli animali hanno mostrato tossicità riproduttiva (vedere paragrafo 5.3). Jaypirca non deve essere usato durante la gravidanza.
Allattamento
Non è noto se pirtobrutinib sia escreto nel latte materno. Il rischio per il lattante non può essere escluso. L'allattamento al seno deve essere interrotto durante il trattamento con Jaypirca e per una settimana dopo l'ultima dose di Jaypirca.
Fertilità
Non esistono dati sull'effetto di pirtobrutinib sulla fertilità umana.
4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Jaypirca altera lievemente la capacità di guidare veicoli e di usare macchinari. Stanchezza, capogiri e astenia sono stati riportati in alcuni pazienti durante il trattamento con Jaypirca e devono essere presi in considerazione quando si valuta la capacità del paziente di guidare veicoli o utilizzare macchinari.
4.8 Effetti indesiderati
Riassunto del profilo di sicurezza
Le reazioni avverse più comuni di qualsiasi grado sono: neutropenia (27,7 %), stanchezza (26,2 %), diarrea (23,8 %), anemia (20,7 %), eruzione cutanea (18,4 %) e contusione (17,8 %).
Le reazioni avverse gravi (grado ≥ 3) più comuni sono: neutropenia (23,9 %), anemia (11,2 %), trombocitopenia (9,7 %) e infezione polmonare (9,0 %).
La frequenza di interruzione del trattamento a causa di reazioni avverse è del 4,2 % e la frequenza di riduzioni della dose a causa di reazioni avverse è del 4,8 %.
Le reazioni avverse più comuni (riportate in più di 2 pazienti) che hanno portato alla riduzione della dose sono neutropenia (2,5 %), eruzione cutanea (0,6 %), diarrea (0,4 %), stanchezza (0,4 %) e trombocitopenia (0,4 %). Le reazioni avverse più comuni (riportate in più di 2 pazienti) che hanno portato all'interruzione della dose sono neutropenia (1,0 %), anemia (1,0 %), infezione polmonare (0,9 %), trombocitopenia (0,7 %) ed eruzione cutanea (0,4 %).
Reazioni avverse gravi associate a Jaypirca si sono verificate nel 19,4 % dei pazienti e le reazioni avverse gravi più comuni (verificatesi in ≥ 1% dei pazienti) sono state infezione polmonare (8,0 %), neutropenia (3,2 %), anemia (2,6 %), fibrillazione atriale/flutter atriale (1,3 %) e infezione delle vie urinarie (1,0 %).
Reazioni avverse fatali sono state osservate nello 0,4 % dei pazienti (3 pazienti) per infezione polmonare, nello 0,3 % dei pazienti (2 pazienti) per emorragia e nello 0,1% dei pazienti (1 paziente) per infezioni delle vie urinarie.
Tabella delle reazioni avverse
La Tabella 1 elenca le reazioni avverse (ADR, adverse drug reaction) associate a Jaypirca usato come monoterapia dai dati degli studi clinici. Le ADR si basano sui dati aggregati di 690 pazienti trattati con Jaypirca in monoterapia alla dose iniziale di 200 mg QD, senza aumento della dose, in uno studio clinico di fase 1/2 e dei pazienti trattati con Jaypirca in monoterapia 200 mg QD in uno studio di fase 3. I pazienti sono stati trattati per MCL, leucemia linfocitica cronica/linfoma a piccoli linfociti (CLL/SLL, small lymphocytic lymphoma) e altri linfomi non-Hodgkin (non-Hodgkin lymphoma, NHL). I pazienti sono stati esposti a Jaypirca per una durata mediana di 12 mesi. Le ADR sono elencate di seguito secondo la classificazione per sistemi e organi secondo MedDRA. I gruppi di frequenza sono definiti dalla seguente convenzione: molto comune (≥ 1/10); comune (≥ 1/100, < 1/10); non comune (≥ 1/1 000, < 1/100); raro (≥ 1/10 000, < 1/1 000); molto raro (< 1/10 000) e non nota (la frequenza non può essere stimata sulla base dei dati disponibili). All'interno di ogni gruppo di frequenza, le ADR sono presentate in ordine decrescente di gravità.
Tabella 1: ADR di pazienti trattati con Jaypirca in monoterapiaa a 200 mg QD
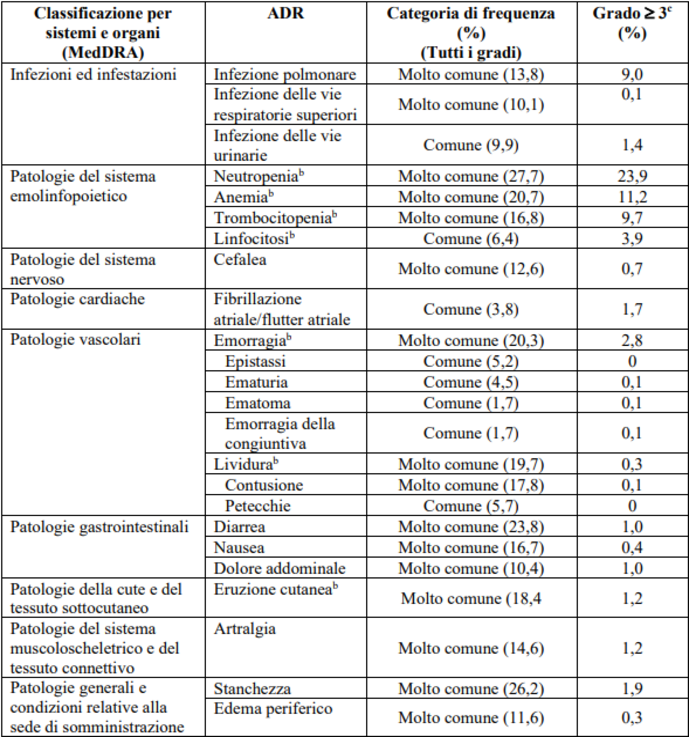
a Le frequenze derivano dall'esposizione a Jaypirca in pazienti con neoplasie maligne a cellule B
b Include più termini di reazione avversa
c Assegnazione del grado di gravità in base ai Common Terminology Criteria for Adverse Events del National Cancer Institute (NCI CTCAE) versione 5.0
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l’autorizzazione del medicinale
è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite l’Agenzia Italiana del Farmaco Sito web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
4.9 Sovradosaggio
Nello studio di fase 1 in cui i pazienti hanno ricevuto dosi ripetute fino a 300 mg una volta al giorno la dose massima tollerata non è stata raggiunta. In studi su volontari sani, non è stata osservata alcuna tossicità correlata alla dose quando è stata somministrata una dose singola massima di 900 mg. Segni e sintomi di sovradosaggio di pirtobrutinib non sono stati stabiliti e non esiste un trattamento specifico per il sovradosaggio di pirtobrutinib.
Nei pazienti che manifestano un sovradosaggio, monitorare attentamente e fornire un trattamento di
supporto appropriato.
5.1 Proprietà farmacodinamiche
Categoria farmacoterapeutica: farmaci antineoplastici, inibitori delle protein chinasi, codice ATC: L01EL05.
Meccanismo d’azione
Pirtobrutinib è un inibitore reversibile, non covalente di BTK. BTK è una proteina della via di segnalazione del recettore per l'antigene delle cellule B (BCR) e del recettore per le citochine. Nelle cellule B, la trasmissione del segnale mediata da BTK determina l'attivazione delle vie necessarie per la proliferazione, il trafficking, la chemiotassi e l'adesione delle cellule B. Pirtobrutinib si lega sia a BTK wild type sia a BTK che presenta mutazioni a carico del sito C481, portando all'inibizione dell'attività della chinasi BTK.
Effetti farmacodinamici
Elettrofisiologia cardiaca
L'effetto di una dose singola di 900 mg di pirtobrutinib sull'intervallo QT corretto (QTc) è stato valutato in uno studio con placebo e controlli positivi in 30 soggetti sani. La dose selezionata è equivalente a circa 2 volte la dose più alta rispetto alle concentrazioni raggiunte allo stato stazionario con la dose raccomandata di 200 mg una volta al giorno. Pirtobrutinib non ha avuto alcun effetto clinicamente significativo sulla variazione dell'intervallo QT corretto per la frequenza cardiaca utilizzando l'intervallo della formula di Fridericia (QTcF) (ovvero > 10 ms) e non vi è stata alcuna relazione tra l'esposizione a pirtobrutinib e la variazione dell'intervallo QTc.
Efficacia e sicurezza clinica
Linfoma a cellule mantellari
L'efficacia di Jaypirca è stata valutata in pazienti adulti con MCL in uno studio clinico multicentrico di fase 1/2, in aperto, a braccio singolo: Studio 18001 (BRUIN). Lo studio comprendeva due parti: la fase 1 di incremento della dose nella quale è stato studiato l'intervallo di dose di pirtobrutinib in monoterapia da 25 mg a 300 mg una volta al giorno, e la fase 2 di espansione della dose. L'obiettivo primario della parte di fase 1 è stato determinare la dose raccomandata di pirtobrutinib per la fase 2, che è risultata essere di 200 mg una volta al giorno, senza identificare una dose massima tollerata.
L'obiettivo primario della parte di fase 2 è stato valutare l'attività antitumorale di pirtobrutinib sulla base del tasso di risposta globale valutato da un comitato di revisione indipendente. I pazienti hanno ricevuto Jaypirca per via orale una volta al giorno fino alla progressione della malattia o a una tossicità inaccettabile.
Lo studio 18001 ha arruolato e trattato un totale di 164 pazienti con diagnosi di MCL e il set di analisi primario (primary analysis set, PAS) per la valutazione dell'efficacia era basato sui primi 90 pazienti con MCL arruolati che non presentavano coinvolgimento noto del sistema nervoso centrale (SNC), che erano stati trattati con un precedente inibitore di BTK, che avevano ricevuto una o più dosi di Jaypirca e che avevano almeno 1 sede di malattia valutabile radiograficamente. L'età mediana era di 70 anni (intervallo: da 46 a 87 anni), l'80 % era di sesso maschile, l'84,4 % era bianco, il 67,8 % aveva un performance status secondo ECOG (Eastern Cooperative Oncology Group) al basale pari a 0 e il 31,1 % aveva un performance status secondo ECOG di 1. I pazienti avevano un numero mediano di 3 linee di terapia precedenti (intervallo: da 1 a 8), e come ragione dell'interruzione della precedente terapia con inibitori di BTK la progressione nell'81,1 % dei pazienti e l'intolleranza nel 13,3 % dei pazienti. Il 95,6 % dei pazienti aveva ricevuto una precedente terapia con anti CD20, l'87,8% chemioterapia, il 18,9 % trapianto di cellule staminali autologhe, il 4,4 % trapianto di cellule staminali allogeniche, il 15,6 % un inibitore di BCL2 e il 4,4 % ha ricevuto una terapia con cellule T esprimenti un recettore antigenico chimerico (CAR-T). Il 38,9 % dei pazienti aveva un coinvolgimento extra nodale e il 26,7 % aveva una massa tumorale di dimensioni maggiori o uguali a 5 cm. Il punteggio semplificato dell'indice prognostico internazionale MCL (MCL International Prognostic Index, sMIPI) era basso nel 22,2 %, intermedio nel 55,6 % e alto nel 22,2 % dei pazienti.
Dei 164 pazienti con MCL arruolati nello Studio 18001, 9 pazienti hanno avuto una riduzione della dose, inclusi 6 responder che sono stati in grado di continuare la terapia e mantenere una risposta duratura in seguito a riduzioni della dose a 150 mg QD (3), 100 mg QD (2) e 50 mg QD (1).
L'efficacia di Jaypirca era basata su una risposta valutata utilizzando i criteri di Lugano del 2014 per i linfomi maligni. I risultati di efficacia per i pazienti che hanno ricevuto almeno un precedente inibitore di BTK e inclusi nel PAS sono riassunti nella Tabella 2. Per i 90 pazienti nel PAS, 79 hanno ricevuto almeno 1 dose di 200 mg QD. Di questi 79 pazienti, 77 hanno iniziato a 200 mg QD, 1 ha aumentato la dose da una dose più bassa e 1 ha ridotto la dose da una dose più alta. Il tempo mediano di trattamento è stato di 5,24 mesi (intervallo: da 0,2 a 39,6 mesi). Tra i 51 responder, il tempo mediano alla risposta è stato di 1,84 mesi (intervallo: da 1,0 a 7,5 mesi).
Sebbene le analisi dei sottogruppi rappresentino un numero limitato di pazienti, sono stati osservati risultati di efficacia clinicamente significativi in importanti ottogruppi, compresi i pazienti che hanno interrotto la precedente terapia con inibitori di BTK a causa di intolleranza o progressione e indipendentemente dal numero e dal tipo di terapie precedenti.
Tabella 2: Riepilogo dei dati di efficacia nello Studio 18001 per i pazienti con MCL che hanno ricevuto almeno un precedente inibitore di BTK
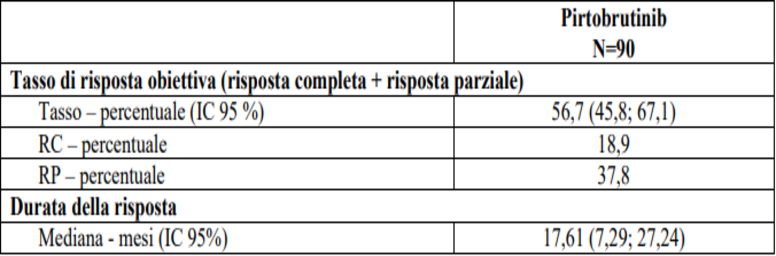
Abbreviazioni: IC = intervallo di confidenza, NS = non stimabile. RC = risposta completa, RP = riposta parziale.
Data di cut-off dei dati: 29 luglio 2022. Il tempo mediano di follow-up per la durata della risposta è stato di 12,68 mesi.
Leucemia linfatica cronica
L'efficacia di Jaypirca è stata valutata nei pazienti con CLL pretrattata con inibitore di BTK in uno studio randomizzato, multicentrico, internazionale, in aperto, con controllo attivo (BRUIN CLL-321, Studio 20020). Lo studio ha arruolato 238 pazienti con CLL/SLL precedentemente trattati con un inibitore di BTK. I pazienti sono stati randomizzati in un rapporto 1:1 ad assumere Jaypirca somministrato per via orale una volta al giorno alla dose di 200 mg fino a progressione di malattia o a tossicità inaccettabile, o ad assumere la terapia scelta dallo sperimentatore:
Idelalisib più un medicinale a base di rituximab (IR): idelalisib 150 mg per via orale due volte al giorno fino a progressione di malattia o a tossicità inaccettabile, in associazione con 8 infusioni di un medicinale a base di rituximab (375 mg/m2 per via endovenosa il giorno 1 del ciclo 1, seguiti da 500 mg/m2 ogni 2 settimane per 4 dosi e poi ogni 4 settimane per 3 dosi), con una durata del ciclo di 28 giorni.
Bendamustina più un medicinale a base di rituximab (BR): bendamustina 70 mg/m2 per via endovenosa (giorno 1 e 2 di ogni ciclo di 28 giorni), in associazione con un medicinale a base 10 di rituximab (375 mg/m2 per via endovenosa il giorno 1 del ciclo 1, poi 500 mg/m2 il giorno 1 dei cicli successivi), per un massimo di 6 cicli.
La randomizzazione è stata stratificata in base allo stato di delezione 17p (sì/no) e all’assunzione di un precedente trattamento con venetoclax (sì/no). Dei 238 pazienti totali, 119 sono stati assegnati alla monoterapia con Jaypirca, 82 a IR e 37 a BR. Dopo la conferma della progressione di malattia, i pazienti randomizzati a IR o BR hanno avuto la possibilità di passare alla monoterapia con Jaypirca.
Le caratteristiche basali erano simili tra i bracci di trattamento. Nel complesso, l'età mediana era di 67 anni (intervallo: da 42 a 90 anni), il 70 % erano maschi e l'81 % erano bianchi. Il performance status secondo ECOG al basale era 0 o 1 nel 93 % dei pazienti e il 44 % dei pazienti aveva malattia in stadio Rai III o IV. Tra i pazienti con test centralizzato disponibile, il 57 % (101 su 176 pazienti) presentava una delezione 17p e/o una mutazione TP53, l'86 % (164 su 190 pazienti) aveva IGHV non mutato e il 65 % (97 su 149) presentava un cariotipo complesso.
I pazienti avevano ricevuto un numero mediano di precedenti linee di terapia pari a 3 (intervallo: da 1 a 13) con il 57 % che era stato sottoposto ad almeno 3 terapie precedenti e il 51 % che era stato sottoposto ad una precedente terapia con BCL2-inibitore. Gli inibitori di BTK più comuni assunti in precedenza sono stati ibrutinib (87 %), acalabrutinib (16 %) e zanubrutinib (7 %). Il 70 % dei pazienti ha interrotto l’inibitore di BTK più recente per malattia refrattaria o progressiva, il 15 % ha interrotto il trattamento per tossicità e il 15 % ha interrotto per altri motivi.
L'efficacia è stata basata sulla sopravvivenza libera da progressione (progression free survival, PFS) di pirtobrutinib in monoterapia rispetto al braccio con la terapia scelta dallo sperimentatore, come valutato da un comitato di revisione indipendente (Independent Review Committee - IRC). Lo studio ha raggiunto l’endpoint primario al momento prespecificato dell'analisi finale per la PFS valutata dall'IRC (cut-off del 29 agosto 2023). In un'analisi aggiornata (cut-off del 29 agosto 2024) con un follow-up mediano di 19,4 mesi (intervallo da 0,03 a 33,3 mesi) per pirtobrutinib e di 17,7 mesi (intervallo da 0,03 a 27,9 mesi) per il braccio con la terapia scelta dallo sperimentatore, è stato osservato un miglioramento della PFS valutata dall'IRC con pirtobrutinib rispetto al braccio con la terapia scelta dallo sperimentatore, in linea con l'analisi primaria. Risultati di efficacia clinicamente significativi a favore di pirtobrutinib sono stati osservati in importanti sottogruppi, inclusi i pazienti che avevano interrotto la precedente terapia con inibitori di BTK a causa di intolleranza o progressione e indipendentemente dal numero e dal tipo di terapie precedenti. I risultati di efficacia sono presentati nella Tabella 3. La curva di Kaplan-Meier per la PFS è mostrata nella Figura 1.
Tabella 3: Risultati di efficacia secondo l’IRC nei pazienti con CLL precedentemente trattati con un inibitore di BTK – Popolazione ITT (Intent to Treat) (Studio 20020)
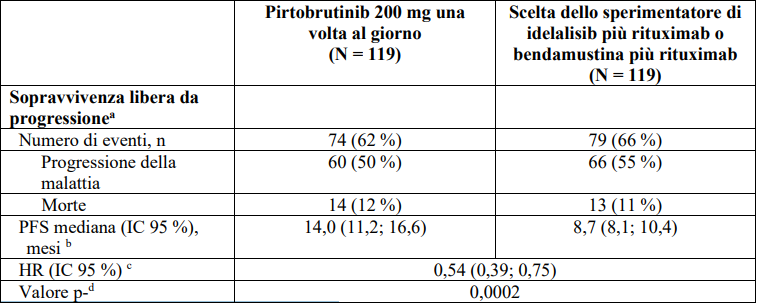
IC, intervallo di confidenza; HR, hazard ratio.
Data di cut-off dei dati 29 agosto 2024
a L'efficacia è stata valutata utilizzando le linee guida dell'International Workshop for Chronic
Lymphocytic Leukemia (iwCLL) del 2018.
b Sulla base di una stima di Kaplan-Meier.
c Basato sul modello stratificato dei rischi proporzionali di Cox.
d Valore p nominale su 2 lati basato su test di rango logaritmico stratificato.
Figura 1: Curva di Kaplan-Meier della PFS valutata dall'IRC nei pazienti con CLL precedentemente trattati con un inibitore di BTK nello studio 20020
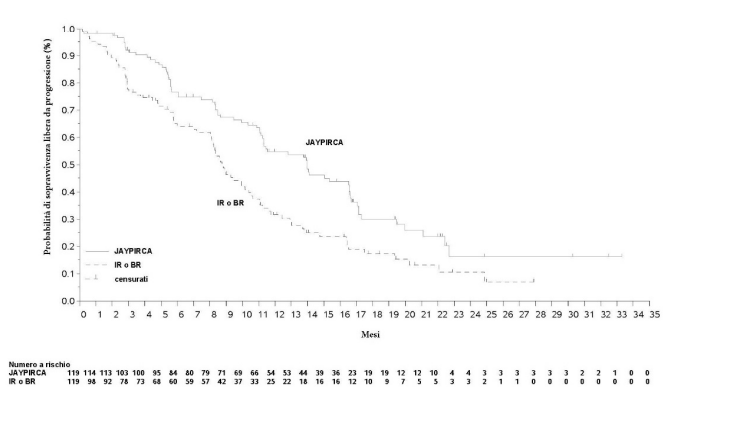
Con un tempo mediano di follow-up di sopravvivenza globale (overall survival, OS) di 20,4 mesi per pirtobrutinib e 19,2 mesi nel braccio a scelta dello sperimentatore, 38 pazienti (32,0 %) nel braccio pirtobrutinib e 32 pazienti (27,0 %) nel braccio a scelta dello sperimentatore sono deceduti. La OS mediana è stata di 29,7 mesi (IC 95 %: 27,1; NS) nel braccio pirtobrutinib e non è stata raggiunta nel braccio scelto dallo sperimentatore. L'HR era 1,090 (IC 95 %: 0,679, 1,749; p = 0,7202). L'analisi della OS potrebbe essere stata influenzata dai 50 pazienti su 119 che sono passati dal braccio scelto dallo sperimentatore a pirtobrutinib.
Popolazione pediatrica
L’Agenzia europea per i medicinali ha previsto l’esonero dall’obbligo di presentare i risultati degli studi con Jaypirca in tutti i sottogruppi della popolazione pediatrica per i tumori maligni a cellule B mature (vedere paragrafo 4.2 per informazioni sull’uso pediatrico).
Approvazione subordinata a condizioni
Questo medicinale è stato autorizzato con procedura “subordinata a condizioni”. Ciò significa che devono essere forniti ulteriori dati su questo medicinale.
L’Agenzia europea per i medicinali esaminerà almeno annualmente le nuove informazioni su questo medicinale e il riassunto delle caratteristiche del prodotto (RCP) verrà aggiornato, se necessario.
5.2 Proprietà farmacocinetiche
La farmacocinetica di pirtobrutinib è stata caratterizzata in soggetti sani e in pazienti con cancro. Le dosi variavano da 25 mg a 300 mg una volta al giorno (da 0,125 a 1,5 volte il dosaggio raccomandato di 200 mg una volta al giorno), fino a dosi singole di 900 mg. Gli aumenti dell'esposizione plasmatica sono stati approssimativamente proporzionali alla dose. Lo stato stazionario è stato raggiunto entro 5 giorni dalla somministrazione una volta al giorno e nei pazienti oncologici il rapporto medio di accumulo [coefficiente di variazione (CV %)] dopo la somministrazione di 200 mg una volta al giorno è stato di 1,63 (26,7 %) in base all'AUC. Tre fattori legati al paziente sono stati attribuiti alle variazioni della farmacocinetica di pirtobrutinib: peso corporeo, albumina sierica ed eGFR assoluto. Si prevede che un aumento del peso corporeo da 70 kg a 120 kg aumenti la clearance di pirtobrutinib del 24 %; si prevede che una diminuzione dell'eGFR assoluto da 90 mL/min a 30 mL/min riduca la clearance di pirtobrutinib del 16%; e si prevede che una diminuzione dell'albumina sierica da 40 g/L a 30 g/L aumenti la clearance di pirtobrutinib del 21 %. È improbabile che questi fattori da soli determinino modifiche significative della farmacocinetica di pirtobrutinib e non sono raccomandati aggiustamenti della dose.
L'AUC e la Cmax medie allo stato stazionario (CV%) sono state rispettivamente di 92 600 h*ng/mL (39 %) e 6 500 ng/mL (25 %) alla dose raccomandata di 200 mg una volta al giorno nei pazienti con cancro.
Al dosaggio raccomandato, pirtobrutinib raggiunge esposizioni farmacocinetiche che possono superare il valore minimo di BTK IC96 e quindi fornire un'inibizione tonica di BTK per tutto il periodo di mono somministrazione giornaliera, indipendentemente dal tasso del turnover intrinseco di BTK.
Assorbimento
La biodisponibilità assoluta di pirtobrutinib dopo una dose singola orale da 200 mg è dell'85,5 % nei soggetti sani. Il tempo mediano per raggiungere il picco di concentrazione plasmatica (tmax) è di circa 2 ore sia nei pazienti oncologici che nei soggetti sani. Non c'è dipendenza dal pH per l'assorbimento.
Effetto del cibo
Un pasto ad alto contenuto di grassi e calorie somministrato a soggetti sani ha ridotto la Cmax di pirtobrutinib del 23 % e ha ritardato il tmax di 1 ora. Non sono stati osservati effetti sull'AUC di pirtobrutinib. Pirtobrutinib può essere assunto con o senza cibo.
Distribuzione
Il volume centrale apparente medio di distribuzione di pirtobrutinib è di 34,2 L nei pazienti oncologici. Il legame alle proteine plasmatiche è del 96 % ed era indipendente dalla concentrazione tra 0,5 e 50 µM. Nel plasma di soggetti sani e di soggetti con compromissione renale severa il legame con le proteine è stato del 96 %. Il rapporto medio sangue/plasma è 0,79.
Biotrasformazione
Il metabolismo epatico è la principale via di clearance di pirtobrutinib. Pirtobrutinib è metabolizzato in diversi metaboliti inattivi da CYP3A4, UGT1A8 e UGT1A9. Non vi è stato alcun impatto clinicamente significativo della modulazione del CYP3A sulle esposizioni a pirtobrutinib.
Pirtobrutinib inibisce CYP2C8, CYP2C9 e CYP3A4 in vitro e inibisce minimamente CYP1A2, CYP2B6, CYP2C19 o CYP2D6 a 60 µM. In vitro pirtobrutinib induce CYP3A4, CYP3A5, CYP2C19 e CYP2B6.
Pirtobrutinib inibisce in maniera minima UGT1A1 in vitro con una CI50 = 18 µM.
Co-somministrazione con substrati/inibitori del trasporto
Studi in vitro hanno indicato che pirtobrutinib è un substrato di P-gp e BCRP.
Pirtobrutinib è un inibitore in vitro di P-gp e BCRP. Negli studi clinici pirtobrutinib ha influito sulla PK della digossina, un substrato della P-gp, e della rosuvastatina, un substrato della BCRP (vedere paragrafo 4.5).
Eliminazione
La clearance apparente media di pirtobrutinib è di 2,05 L/h con un'emivita effettiva di circa 19,9 ore. Dopo una dose singola di pirtobrutinib 200 mg radiomarcato in soggetti sani, il 37% della dose è stato recuperato nelle feci (il 18 % invariato) e il 57% nelle urine (il 10 % invariato).
Popolazioni speciali
Età, sesso, popolazione e peso corporeo
Sulla base di un'analisi farmacocinetica di popolazione in pazienti affetti da cancro, età (intervallo 227-95 anni), popolazione, sesso e peso corporeo (intervallo 35,7-152,5 kg) non hanno avuto effetti clinicamente significativi sull'esposizione a pirtobrutinib.
Compromissione renale
In un'analisi farmacocinetica di popolazione di pazienti oncologici, pazienti con compromissione renale lieve (eGFR da 60 a < 90 mL/min) o moderata (eGFR da 30 a < 60 mL/min), la clearance di pirtobrutinib era inferiore dal 16 % al 27 % rispetto alla clearance nei pazienti con funzione renale normale, con conseguente esposizione attesa di AUC = 94 100 ng*h/mL e Cmax = 6 680 ng/mL in pazienti con compromissione renale lieve (16-19% più alta rispetto ai pazienti con funzione renale normale) e AUC = 108 000 ng*h/mL e Cmax = 7 360 ng/mL in pazienti con compromissione renale moderata (dal 28 al 36 % più alta rispetto ai pazienti con funzionalità renale normale).
In uno studio di farmacologia clinica su volontari altrimenti sani, la clearance apparente era inferiore del 35 % in quattro partecipanti con compromissione renale grave (eGFR da 15 a < 30 mL/min) rispetto a otto partecipanti con funzionalità renale normale (eGFR ≥ 90 mL/min), con conseguenti esposizioni di AUC0 inf = 115 000 ng*h/mL e Cmax = 2 980 ng/mL (rispettivamente 62 % più alta e 7% più bassa, rispetto alla normale funzionalità renale).
I pazienti con malattia renale allo stadio terminale sottoposti a dialisi non sono stati studiati (vedere paragrafo 4.2).
Compromissione epatica
Non sono state rilevate differenze clinicamente significative nella farmacocinetica di pirtobrutinib per qualsiasi grado di compromissione epatica (secondo Child-Pugh A, B e C o qualsiasi valore di bilirubina totale e qualsiasi valore di AST). In uno studio dedicato sulla compromissione epatica, l'AUC e la Cmax medie di pirtobrutinib erano simili tra i soggetti con compromissione epatica lieve (Child Pugh A) e i soggetti con funzionalità epatica normale. Nei soggetti con compromissione epatica moderata (Child-Pugh B) l'AUC era inferiore del 15 % rispetto alla funzionalità epatica normale e la Cmax era simile. Nei soggetti con compromissione epatica severa (Child Pugh C) l'AUC di pirtobrutinib era inferiore del 21 % e la Cmax media era inferiore del 24 % rispetto ai soggetti con funzionalità epatica normale. La frazione non legata (fu – fraction unbound) per pirtobrutinib nei soggetti è generalmente aumentata con l'aumentare della severità della compromissione epatica.
Pertanto, dopo aver corretto i parametri di esposizione farmacocinetica di pirtobrutinib con la fu, non è stata osservata alcuna differenza clinicamente significativa nei parametri di esposizione farmacocinetica di pirtobrutinib non legato (AUCu e Cmax,u) tra soggetti con qualsiasi grado di compromissione epatica e funzionalità epatica normale.
Popolazione pediatrica
Non sono stati condotti studi di farmacocinetica con pirtobrutinib in pazienti di età inferiore a 18 anni.
5.3 Dati preclinici di sicurezza
Negli studi condotti con dosi ripetute sono state osservate una ridotta risposta anticorpale dipendente dalle cellule T nei ratti (a un'esposizione umana di 0,69 volte alla dose raccomandata di 200 mg basata sull'AUC) e lesioni corneali da minime a lievi nei cani (a un'esposizione umana di 0,42 volte). Necrosi vascolare da lieve a moderata e infiammazione vascolare/perivascolare dei grandi vasi sanguigni polmonari sono state osservate solo nei ratti. Questi effetti si sono verificati a livelli di esposizione clinicamente rilevanti.
Genotossicità/cancerogenicità
Pirtobrutinib non è risultato mutageno in un test di mutagenicità batterica (Ames). Pirtobrutinib è risultato aneugenico in due test del micronucleo in vitro utilizzando linfociti del sangue periferico umano. Pirtobrutinib non ha avuto alcun effetto in un test del micronucleo del midollo osseo di ratto in vivo a dosi fino a 2 000 mg / kg (dose singola), esposizione circa 11 volte più alta (considerando il valore della Cmax non legata in animali femmine) rispetto all'esposizione umana a 200 mg.
Non sono stati condotti studi di cancerogenicità con pirtobrutinib.
Embriotossicità/teratogenicità
Negli studi sulla riproduzione animale, la somministrazione di pirtobrutinib a ratte gravide durante l'organogenesi ha determinato una riduzione del peso fetale, mortalità embrio-fetale e malformazioni fetali a esposizioni materne di 3,0 volte l'esposizione umana alla dose raccomandata di 200 mg in base all'AUC.
Tossicità riproduttiva
Non sono stati condotti studi di fertilità con pirtobrutinib. In studi di tossicità a dose ripetuta della durata fino a 3 mesi, pirtobrutinib non ha avuto effetti sugli organi riproduttivi maschili a 0,69 volte e 0,42 volte l’esposizione umana rispettivamente nei ratti e nei cani, alla dose raccomandata di 200 mg in base all'AUC. Pirtobrutinib non ha avuto alcun effetto sugli organi riproduttivi femminili a 4,0 volte e 0,42 volte l’esposizione rispettivamente nei ratti e nei cani.
6.1 Elenco degli eccipienti
Nucleo della compressa
Ipromellosa acetato succinato
Cellulosa microcristallina
Lattosio monoidrato
Sodio croscarmelloso
Stearato di magnesio
Silice colloidale idrata
Film di rivestimento
Ipromellosa
Biossido di titanio
Triacetina
Indaco carminio (E132)
6.2 Incompatibilità
Non pertinente.
6.3 Periodo di validità
3 anni
6.4 Precauzioni particolari per la conservazione
Questo medicinale non richiede alcuna condizione particolare di conservazione.
6.5 Natura e contenuto del contenitore
Jaypirca 50 mg compresse rivestite con film
Blister in cloruro di polivinile/policlorotrifluoroetilene sigillati con un foglio di alluminio in confezioni da 28, 30 o 84 compresse rivestite con film.
Jaypirca 100 mg compresse rivestite con film
Blister in cloruro di polivinile/policlorotrifluoroetilene sigillati con un foglio di alluminio in confezioni da 28, 30, 56, 60, 84 o 168 compresse rivestite con film.
È possibile che non tutte le confezioni siano commercializzate.
6.6 Precauzioni particolari per lo smaltimento
Il medicinale non utilizzato e i rifiuti derivati da tale medicinale devono essere smaltiti in conformità
alla normativa locale vigente.
Eli Lilly Nederland B.V.
Papendorpseweg 83
3528 BJ Utrecht
Paesi Bassi
EU/1/23/1738/001
EU/1/23/1738/002
EU/1/23/1738/003
EU/1/23/1738/004
EU/1/23/1738/005
EU/1/23/1738/006
EU/1/23/1738/007
EU/1/23/1738/008
EU/1/23/1738/009
Data della prima autorizzazione: 30 ottobre 2023
Data del rinnovo più recente: 16 agosto 2024
28 marzo 2025
Informazioni più dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia europea per i medicinali, https://www.ema.europa.eu.