Per segnalare un effetto indesiderato o un reclamo su un prodotto Lilly, si prega di non utilizzare questo sito web. La segnalazione delle reazioni avverse è estremamente importante. Gli operatori sanitari e i pazienti possono riportare una reazione avversa o un reclamo di un prodotto sul sito di AIFA.
Retsevmo® (selpercatinib)
Medicinale sottoposto a monitoraggio addizionale. Ciò permetterà la rapida identificazione di nuove informazioni sulla sicurezza. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta. Vedere paragrafo 4.8 per informazioni sulle modalità di segnalazione delle reazioni avverse.Retsevmo 40 mg capsule rigide
Retsevmo 80 mg capsule rigide
Retsevmo 40 mg capsule rigide
Ciascuna capsula rigida contiene 40 mg di selpercatinib.
Retsevmo 80 mg capsule rigide
Ciascuna capsula rigida contiene 80 mg di selpercatinib.
Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
Capsule rigide.
Retsevmo 40 mg capsule rigide
Capsule grigio opaco, 6 x 18 mm (dimensione 2), con impresso in inchiostro nero “Lilly”, “3977” e “40 mg”.
Retsevmo 80 mg capsule rigide
Capsule blu opaco, 8 x 22 mm (dimensione 0), con impresso in inchiostro nero “Lilly”, “2980” e “80 mg”.
4.1 Indicazioni terapeutiche
Retsevmo come monoterapia è indicato nel trattamento di adulti con:
- cancro del polmone non a piccole cellule (non-small cell lung cancer, NSCLC) avanzato RET fusione-positivo non precedentemente trattati con un inibitore di RET. Per la selezione dei pazienti basata sui biomarcatori, vedere paragrafo 4.2
Retsevmo come monoterapia è indicato nel trattamento di adulti e pazienti pediatrici di età pari o superiore a 2 anni con:
- cancro della tiroide avanzato RET fusione-positivo che sono refrattari allo iodio radioattivo (se lo iodio radioattivo è appropriato). Per la selezione dei pazienti basata sui biomarcatori, vedere paragrafo 4.2
- cancro midollare della tiroide (medullary thyroid cancer, MTC) avanzato con mutazione di RET. Per la selezione dei pazienti basata sui biomarcatori, vedere paragrafo 4.2.
- tumori solidi avanzati RET fusione-positivi, quando le opzioni terapeutiche non mirate a RET forniscono un beneficio clinico limitato o sono state esaurite (vedere paragrafi 4.4 e 5.1). Per la selezione dei pazienti basata sui biomarcatori, vedere paragrafo 4.2
4.2 Posologia e modo di somministrazione
La terapia con Retsevmo deve essere iniziata e supervisionata da medici esperti nell’uso di terapie anti-tumorali.
Selezione del paziente
Prima dell’inizio del trattamento con Retsevmo, la presenza di una mutazione (MTC) o di una fusione del gene RET (tutti gli altri tipi di tumore) deve essere valutata con un IVD (dispositivo medico diagnostico in vitro) con marchio CE per il corrispondente scopo previsto. Se l'IVD con marchio CE non è disponibile, deve essere valutato con un test alternativo validato .
Posologia
La dose raccomandata di Retsevmo nei pazienti di età pari o superiore a 12 anni in base al peso corporeo è:
- meno di 50 kg: 120 mg due volte al giorno
- 50 kg o più: 160 mg due volte al giorno.
La dose raccomandata di Retsevmo per i pazienti pediatrici di età compresa tra 2 e meno di 12 anni si basa sulle seguenti categorie di superficie corporea (BSA) (Tabella 1):
Tabella 1 Dose raccomandata per i pazienti pediatrici di età compresa tra 2 e meno di 12 anni

Retsevmo non è raccomandato per pazienti pediatrici con superficie corporea inferiore a 0,45 m2.
Le compresse sono disponibili per i pazienti pediatrici che non sono in grado di deglutire le capsule (fare riferimento all’RCP delle compresse).
Se un paziente vomita o salta una dose, deve essere istruito a prendere la dose successiva all’orario programmato; non deve essere assunta una dose aggiuntiva.
Il trattamento deve essere continuato fino a progressione di malattia o tossicità inaccettabile.
La dose attuale di selpercatinib deve essere ridotta del 50% se co-somministrata con un forte inibitore del CYP3A. Se viene sospeso l’inibitore del CYP3A, la dose di selpercatinib deve essere aumentata (dopo 3-5 emivite dell’inibitore) alla dose che era utilizzata prima di iniziare l’inibitore.
Aggiustamenti della dose
La gestione di alcune reazioni avverse può richiedere l’interruzione e/o la riduzione della dose. Le modifiche di dose di Retsevmo sono riassunte nella tabella 2 e nella tabella 3.
Tabella 2 Modifiche della dose raccomandate per Retsevmo sulla base del peso corporeo e della superficie corporea, in caso di reazioni avverse

*Interrompere definitivamente selpercatinib nei pazienti che non sono in grado di tollerare tre riduzioni della dose
Le modifiche della dose nei pazienti pediatrici per reazioni avverse devono essere determinate in base alla dose attuale e a un approccio di modifica graduale del livello di dose, come quello descritto per gli adulti nelle reazioni avverse discusse di seguito (Tabella 3), salvo diversa indicazione.
Tabella 3 Modifiche della dose raccomandate a causa di reazioni avverse


Particolari popolazioni
Anziani
Non è richiesto nessun aggiustamento di dose sulla base dell’età (vedere paragrafo 5.2).
Non sono state osservate differenze complessive negli eventi avversi emersi durante il trattamento o nell’efficacia di selpercatinib tra i pazienti che avevano un’età ≥ 65 anni e nei pazienti più giovani. Per pazienti ≥ 75 anni sono disponibili dati limitati.
Compromissione renale
Non sono necessari aggiustamenti di dose nei pazienti con compromissione renale lieve, moderata o severa. Non ci sono dati per pazienti con nefropatia allo stadio terminale o in pazienti in dialisi (paragrafo 5.2).
Compromissione epatica
È importante lo stretto monitoraggio dei pazienti con funzionalità epatica compromessa. Non sono richiesti aggiustamenti di dose per pazienti con compromissione della funzionalità epatica lieve (Child-Pugh classe A) o moderata (Child-Pugh classe B). Adulti con compromissione della funzionalità epatica severa (Child-Pugh classe C) devono ricevere una dose di selpercatinib di 80 mg due volte al giorno (paragrafo 5.2). L’uso di selpercatinib non è stato studiato in pazienti di età inferiore a 18 anni con compromissione epatica. Il trattamento nei pazienti di età inferiore a 18 anni con compromissione epatica grave deve essere iniziato con cautela e gli aggiustamenti della dose devono essere guidati dal giudizio clinico e dalla tollerabilità individuale del paziente. Per i pazienti pediatrici di età inferiore a 12 anni con compromissione epatica grave, la dose iniziale raccomandata è riportata nella Tabella 4.
Tabella 4 Dose iniziale raccomandata per pazienti pediatrici di età inferiore a 12 anni con compromissione epatica grave

Popolazione pediatrica
Retsevmo non deve essere usato in bambini di età inferiore ai 2 anni poiché non sono disponibili dati.
Retsevmo si può usare a partire dall’età di 2 anni per il trattamento di pazienti con MTC con mutazione di RET, cancro della tiroide RET fusione-positivo e tumori solidi RET-fusione positivi (vedere paragrafi 4.4 e 5.1). I pazienti di età pari o superiore a 12 anni devono ricevere la dose a seconda del peso corporeo (vedere paragrafo 4.2). I pazienti di età compresa tra 2 e meno di 12 anni devono ricevere la dose secondo la superficie corporea (vedere paragrafo 4.2). Sulla base dei risultati di uno studio preclinico (vedere paragrafo 5.3), nei pazienti adolescenti devono essere monitorate le cartilagini di accrescimento aperte (non saldate). Deve essere presa in considerazione l’interruzione o la sospensione della dose sulla base della severità di qualsiasi anomalia delle cartilagini di accrescimento e sulla base di una valutazione rischio-beneficio individuale.
Modo di somministrazione
Retsevmo è per uso orale.
Le capsule devono essere deglutite intere (i pazienti non devono aprirle, schiacciarle o masticarle prima di deglutirle) e possono essere assunte con o senza cibo. Le compresse sono disponibili per i pazienti pediatrici che non sono in grado di deglutire le capsule (fare riferimento all’RCP delle compresse).
I pazienti devono assumere le dosi approssimativamente alla stessa ora ogni giorno.
Retsevmo deve essere accompagnato da un pasto se si usa un inibitore della pompa protonica (vedere paragrafo 4.5).
Retsevmo deve essere somministrato 2 ore prima o 10 ore dopo l’assunzione di antagonisti del recettore H2 (vedere paragrafo 4.5).
4.3 Controindicazioni
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
4.4 Avvertenze speciali e precauzioni d’impiego
Efficacia in tutti i tipi di tumore
Il beneficio di selpercatinib è stato stabilito in studi a braccio singolo che hanno coinvolto un campione relativamente piccolo di pazienti i cui tumori presentavano fusioni del gene RET. Gli effetti favorevoli di selpercatinib sono stati dimostrati sulla base del tasso di risposta obiettiva e della durata della risposta in un numero limitato di tipi di tumore. L'effetto può essere quantitativamente diverso a seconda del tipo di tumore, così come delle alterazioni genomiche concomitanti (vedere paragrafo 5.1). Per questi motivi, selpercatinib deve essere usato solo se non ci sono opzioni terapeutiche per le quali è stato stabilito un beneficio clinico, o se tali opzioni terapeutiche sono state esaurite (cioè, nessuna opzione terapeutica soddisfacente).
Malattia polmonare interstiziale (ILD)/polmonite
Sono stati segnalati casi gravi, pericolosi per la vita o fatali di ILD/polmonite in pazienti trattati con selpercatinib (vedere paragrafo 4.8). I pazienti devono essere monitorati per i sintomi polmonari indicativi di ILD/polmonite. Selpercatinib deve essere sospeso e i pazienti devono essere tempestivamente esaminati per ILD se presentano sintomi respiratori acuti o in peggioramento che possono essere indicativi di ILD (ad es. dispnea, tosse e febbre) e trattati come appropriato dal punto di vista medico. In base alla gravità dell'ILD/polmonite, la dose di selpercatinib deve essere interrotta, ridotta o definitivamente sospesa (vedere paragrafo 4.2).
Aumenti di alanina aminotransferasi (ALT)/aspartato aminotransferasi (AST)
Aumenti di ALT di Grado ≥ 3 e aumenti di AST di Grado ≥ 3 sono stati segnalati in pazienti trattati con selpercatinib (vedere paragrafo 4.8). ALT e AST devono essere monitorate prima di iniziare la terapia con selpercatinib, ogni 2 settimane durante i primi 3 mesi di trattamento, una volta al mese nei successivi 3 mesi di trattamento o come clinicamente indicato. Sulla base dell’innalzamento dei livelli di ALT e AST, selpercatinib può richiedere modifiche del dosaggio (vedere paragrafo 4.2).
Ipertensione
Nei pazienti che hanno ricevuto selpercatinib è stata segnalata ipertensione (vedere paragrafo 4.8). La pressione arteriosa deve essere controllata prima di iniziare il trattamento con selpercatinib, monitorata durante il trattamento con selpercatinib e, se necessario, trattata con la terapia antipertensiva standard. In base all’aumento dei livelli di pressione arteriosa, selpercatinib può richiedere modifiche della dose (vedere paragrafo 4.2). Selpercatinib deve essere interrotto definitivamente se l’ipertensione clinicamente significativa non può essere controllata con terapia antipertensiva.
Prolungamento dell’intervallo QT
Nei pazienti che hanno ricevuto selpercatinib è stato segnalato un prolungamento dell’intervallo QT (vedere paragrafo 5.1). Selpercatinib deve essere usato con cautela in pazienti con condizioni come la sindrome congenita del QT lungo o sindrome del QT lungo acquisita o altre condizioni cliniche che predispongono ad aritmia.
I pazienti devono avere un intervallo QTcF ≤ 470 ms (≤ 440 ms nei pazienti di età inferiore a 12 anni) ed elettroliti sierici entro i limiti di norma prima dell’inizio del trattamento con selpercatinib. Elettrocardiogramma ed elettroliti sierici devono essere monitorati in tutti i pazienti dopo 1 settimana di trattamento con selpercatinib, almeno una volta al mese per i primi 6 mesi o altrimenti come clinicamente indicato, modificando la frequenza sulla base dei fattori di rischio inclusi diarrea, vomito e/o nausea. Ipokaliemia, ipomagnesemia e ipocalcemia devono essere corrette prima di iniziare selpercatinib e durante il trattamento. Monitorare l’intervallo QT tramite ECG, con più frequenza nei pazienti che richiedono trattamento con medicinali concomitanti noti per prolungare l’intervallo QT.
Selpercatinib potrebbe richiedere interruzione o modifiche della dose (vedere paragrafo 4.2).
Ipotiroidismo
Nei pazienti che hanno ricevuto selpercatinib è stato segnalato ipotiroidismo (vedere paragrafo 4.8). In tutti i pazienti è raccomandata la misurazione al basale, dei parametri di laboratorio della funzionalità tiroidea. I pazienti con ipotiroidismo preesistente, devono essere trattati secondo la pratica medica standard prima di iniziare il trattamento con selpercatinib. Tutti i pazienti devono essere tenuti sotto stretta osservazione per segni e sintomi di disfunzione tiroidea durante il trattamento con selpercatinib. La funzionalità tiroidea deve essere monitorata periodicamente durante il trattamento con selpercatinib. I pazienti che sviluppano disfunzione tiroidea devono essere trattati secondo la pratica medica standard, tuttavia i pazienti potrebbero avere una risposta insufficiente alla sostituzione con levotiroxina (T4) poiché selpercatinib può inibire la conversione della levotiroxina a triiodotironina (T3) e può essere necessaria una supplementazione con liotironina (vedere paragrafo 4.5).
Forti induttori del CYP3A
L’uso concomitante di forti induttori del CYP3A4 deve essere evitato a causa del rischio di diminuire l’efficacia di selpercatinib (vedere paragrafo 4.5).
Donne in età fertile/contraccezione nelle donne e negli uomini
Le donne in età fertile devono usare misure contraccettive altamente efficaci durante il trattamento e per almeno una settimana dopo l’ultima dose di selpercatinib. Uomini con compagne in età fertile devono usare misure contraccettive efficaci durante il trattamento e per almeno una settimana dopo l’ultima dose di selpercatinib (vedere paragrafo 4.6).
Fertilità
Sulla base di evidenze di sicurezza non cliniche, la fertilità maschile e femminile potrebbe essere compromessa dal trattamento con Retsevmo (vedere paragrafi 4.6 e 5.3). Uomini e donne devono chiedere consiglio sulle modalità per preservare la fertilità prima del trattamento.
Ipersensibilità
È stata segnalata ipersensibilità in pazienti che hanno ricevuto selpercatinib, e la maggioranza di eventi è stata osservata nei pazienti con NSCLC trattati in precedenza con immunoterapia anti PD-1/PD-L1 (vedere paragrafo 4.8). Segni e sintomi di ipersensibilità hanno incluso febbre, eruzione cutanea e artralgia o mialgia con diminuzione concomitante delle piastrine o aumento delle aminotransferasi.
Sospendere selpercatinib se si manifesta ipersensibilità, e iniziare il trattamento con steroidi. Sulla base del grado delle reazioni di ipersensibilità, selpercatinib può richiedere modifiche di dose (vedere paragrafo 4.2). Gli steroidi devono essere continuati fino a che il paziente raggiunge la dose target e poi diminuiti. Interrompere definitivamente selpercatinib nel caso di ipersensibilità ricorrente.
Emorragie
Eventi gravi, inclusa emorragia letale, sono stati segnalati in pazienti che hanno ricevuto selpercatinib (vedere paragrafo 4.8).
Interrompere definitivamente selpercatinib nei pazienti con emorragia potenzialmente letale o ricorrente severa (vedere paragrafo 4.2).
Sindrome da lisi tumorale (Tumour Lysis Syndrome,TLS)
In pazienti trattati con selpercatinib sono stati osservati casi di TLS. Fattori di rischio per TLS
includono elevato carico tumorale, insufficienza renale cronica preesistente, oliguria, disidratazione, ipotensione e urina acida. Questi pazienti devono essere attentamente monitorati, trattati come clinicamente indicato e dovrebbe essere considerata una profilassi appropriata che includa l’idratazione.
Epifisiolisi della testa del femore nei pazienti pediatrici
L'epifisiolisi della testa del femore è stata riportata in pazienti pediatrici (< 18 anni di età) trattati con selpercatinib (vedere paragrafo 4.8). I pazienti devono essere monitorati per i sintomi indicativi di epifisiolisi della testa del femore e trattati come appropriato dal punto di vista medico e chirurgico.
Gravi reazioni avverse cutanee (Severe cutaneous adverse reactions, SCARs)
La sindrome di Stevens-Johnson (SJS), che può essere pericolosa per la vita o fatale, è stata segnalata in ssociazione al trattamento con selpercatinib (vedere paragrafo 4.8). I pazienti devono essere informati dei segni delle gravi reazioni avverse cutanee e devono consultare immediatamente il proprio medico al momento dell'osservazione di segni o sintomi indicativi. Se compaiono segni e sintomi indicativi di queste reazioni, selpercatinib deve essere sospeso immediatamente e deve essere preso in considerazione un rattamento alternativo (se opportuno). Se, con l'uso di selpercatinib, il paziente ha viluppato una grave reazione avversa utanea come la SJS, il trattamento con selpercatinib non deve essere ripreso in alcun momento in questo paziente.
4.5 Interazioni con altri medicinali ed altre forme d’interazione
Effetti di altri medicinali sulla farmacocinetica di selpercatinib
Il metabolismo di selpercatinib avviene attraverso il CYP3A4. Perciò, i medicinali in grado di influenzare l’attività dell’enzima CYP3A4 possono alterare la farmacocinetica di selpercatinib.
In vitro selpercatinib è un substrato per la glicoproteina P (P-glycoprotein, P-gp) e per la proteina di resistenza del cancro della mammella (Breast Cancer Resistance Protein, BCRP). Tuttavia questi trasportatori non sembrano limitare l’assorbimento orale di selpercatinib, poiché la sua biodisponibilità è del 73% e la sua esposizione è aumentata in maniera minima dalla co-somministrazione di rifampicina, inibitore della P-gp (incremento di circa il 6,5% e 19% rispettivamente nell’AUC0-24 e Cmax di selpercatinib).
Agenti che possono aumentare la concentrazione plasmatica di selpercatinib
La co-somministrazione di una dose singola di 160 mg di selpercatinib con itraconazolo, un forte inibitore del CYP3A, ha aumentato la Cmax e AUC di selpercatinib rispettivamente del 30% e 130%, rispetto a selpercatinib somministrato da solo. Se devono essere co-somministrati forti inibitori del CYP3A e/o P-gp, inclusi a titolo esemplificativo, ketoconazolo, itraconazolo, voriconazolo, ritonavir, saquinavir, telitromicina, posaconazolo e nefazodone, la dose di selpercatinib deve essere ridotta (vedere paragrafo 4.2).
Agenti che possono diminuire la concentrazione plasmatica di selpercatinib
La co-somministrazione di rifampicina, un forte induttore del CYP3A4 ha portato ad una diminuzione di circa l’87% ed il 70% rispettivamente dell’AUC e Cmax di selpercatinib, rispetto a selpercatinib somministrato da solo. Di conseguenza, l’uso concomitante di forti induttori del CYP3A4, inclusi a titolo esemplificativo, carbamazepina, fenobarbital, fenitoina, rifabutina, rifampicina ed Erba di San Giovanni (Hypericum perforatum), deve essere evitato.
Effetti di selpercatinib sulla farmacocinetica di altri medicinali (aumenti nella concentrazione plasmatica)
Substrati sensibili del CYP2C8
Selpercatinib ha aumentato la Cmax e AUC di repaglinide (un substrato del CYP2C8) rispettivamente di circa il 91% e il 188%. Perciò, la co-somministrazione con substrati sensibili del CYP2C8 (per es. amodiachina, cerivastatina, enzalutamide, paclitaxel, repaglinide, torasemide, sorafenib, rosiglitazone, buprenorfina, selexipag, dasabuvir e montelukast) deve essere evitata.
Substrati sensibili del CYP3A4
Selpercatinib ha aumentato la Cmax e AUC di midazolam (un substrato CYP3A4) rispettivamente di circa il 39% e 54%. Perciò, l’uso concomitante di substrati sensibili al CYP3A4 (per es. alfentanil, avanafil, buspirone, conivaptan, darifenacin, darunavir, ebastina, lomitapide, lovastatina, midazolam, naloxegol, nisolpidina, saquinavir, simvastatina, tipranavir, triazolam, vardenafil) deve essere evitato.
Co-somministrazione di medicinali che agiscono sul pH gastrico
Selpercatinib ha una solubilità pH-dipendente, con una ridotta solubilità a valori di pH più alti. Non sono state osservate differenze clinicamente significative nella farmacocinetica di selpercatinib quando co-somministrato con dosi giornaliere multiple di ranitidina (antagonista del recettore H2)assunte 2 ore dopo la dose di selpercatinib.
Co-somministrazione con inibitori di pompa protonica
La co-somministrazione con dosi giornaliere multiple di omeprazolo (un inibitore della pompa protonica) ha diminuito l’AUC0-INF e la Cmax di selpercatinib, quando selpercatinib è stato somministrato a digiuno. La co-somministrazione con dosi giornaliere multiple di omeprazolo non ha cambiato in maniera significativa l’AUC0-INF e Cmax di selpercatinib, quando Retsevmo è stato somministrato insieme al cibo.
Co-somministrazione con medicinali che sono substrati di trasportatori
Selpercatinib inibisce la proteina renale di estrusione multifarmaco e di tossine 1 (multidrug and toxin extrusion protein 1, MATE1). In vivo, può avvenire l’interazione di selpercatinib con substrati clinicamente rilevanti di MATE1, come la creatinina (vedere paragrafo 5.2).
Selpercatinib è un inibitore in vitro della P-gp e BCRP. In vivo, selpercatinib ha aumentato la Cmax e l’AUC di dabigatran, un substrato della P-gp, rispettivamente del 43% e 38%. Pertanto, bisogna prestare attenzione quando si assume un substrato sensibile della P-gp (per es. fexofenadina, dabigatran etexilato, colchicina, saxagliptin) e in particolare quelli con un indice terapeutico ristretto (per es. digossina) (vedere paragrafo 5.2).
In vivo, selpercatinib ha aumentato la Cmax e l’AUC di rosuvastatina, un substrato di BCRP, rispettivamente del 71 % e dell’80 %. Bisogna prestare attenzione quando si assume un substrato di BCRP (ad es., rosuvastatina, prazosina) (vedere paragrafo 5.2).
Medicinali che possono essere meno efficaci quando somministrati con selpercatinib
Selpercatinib potrebbe inibire la D2 deiodinasi e quindi diminuire la conversione di levotiroxina (T4) a triiodotironina (T3). I pazienti potrebbero quindi avere una risposta insufficiente alla sostituzione con levotiroxina e potrebbe essere necessaria un’integrazione con liotironina (vedere paragrafo 4.4).
Popolazione pediatrica
Sono stati effettuati studi d’interazione solo negli adulti.
4.6 Fertilità, gravidanza e allattamento
Donne in età fertile/Contraccezione nelle donne e negli uomini
Le donne in età fertile devono utilizzare misure contraccettive altamente efficaci durante il trattamento e per almeno una settimana dopo l'ultima dose di selpercatinib. Gli uomini con compagne in età fertile devono usare misure contraccettive efficaci durante il trattamento e per almeno una settimana dopo l’ultima dose di selpercatinib.
Gravidanza
I dati relativi all’uso di selpercatinib in donne in gravidanza non esistono. Gli studi sugli animali hanno mostrato una tossicità riproduttiva (vedere paragrafo 5.3). Retsevmo non è raccomandato durante la gravidanza e in donne in età fertile che non usano misure contraccettive. Deve essere usato durante la gravidanza solo se il beneficio potenziale giustifica il potenziale rischio sul feto.
Allattamento
Non è noto se selpercatinib sia escreto nel latte materno. Il rischio per i neonati/lattanti allattati al seno non può essere escluso. L’allattamento con latte materno deve essere interrotto durante il trattamento con Retsevmo e per almeno una settimana dopo l'ultima dose.
Fertilità
Non sono disponibili dati sugli effetti di selpercatinib sulla fertilità negli esseri umani. Sulla base di studi sugli animali è probabile che la fertilità femminile e maschile sia compromessa dal trattamento con Retsevmo (vedere paragrafo 5.3). Uomini e donne devono chiedere consiglio sulle modalità per preservare la fertilità prima del trattamento.
4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Retsevmo può alterare lievemente la capacità di guidare veicoli e di usare macchinari.
I pazienti devono essere avvertiti di prestare attenzione mentre guidano o usano macchinari nel caso manifestino stanchezza o capogiri durante il trattamento con Retsevmo (vedere paragrafo 4.8).
4.8 Effetti indesiderati
Riassunto del profilo di sicurezza
Viene riassunta la frequenza integrata di reazioni avverse al farmaco (ADR) riportate, nei pazienti trattati con selpercatinib da uno studio di fase 1/2 in aperto, multicentrico, con aumento della dose (LIBRETTO-001) e da due studi comparativi di fase 3 randomizzati multicentrici in aperto (LIBRETTO-431 e LIBRETTO-531). Le ADR gravi più comuni (≥ 1,0%) sono infezione polmonare (5,3%), emorragia (2,4%), dolore addominale (2,1%), sodio ematico diminuito (2,0%), diarrea (1,5%), ipersensibilità (1,4%), vomito (1,3%), creatinina ematica aumentata ( 1,3%), piressia (1,3%), infezioni del tratto urinario (1,3%), ALT aumentata (1,0%), AST aumentata (1,0%).
Retsevmo è stato interrotto definitivamente a causa di eventi avversi emersi durante il trattamento nell’8,8% dei pazienti, indipendentemente dall’attribuzione. Le ADR più comuni che hanno portato all’interruzione definitiva del trattamento (3 o più pazienti) sono state ALT aumentata (0,7%), stanchezza (0,5%), AST aumentata (0,4%), bilirubina ematica aumentata (0,3%), infezione polmonare (0,3%), trombocitopenia (0,3%), emorragia (0,3%) e ipersensibilità (0,3%).
Tabella delle reazioni avverse
La frequenza integrata e la gravità delle ADR segnalate nei pazienti trattati con selpercatinib negli studi LIBRETTO-001, LIBRETTO-431 e LIBRETTO-531 sono elencate nella tabella 5.
Le ADR sono classificate in base alla classificazione per sistemi e organi e frequenza secondo MedDRA. Per la classificazione della frequenza è stata usata la seguente convenzione: molto comune (≥ 1/10); comune (≥ 1/100, < 1/10); non comune (≥ 1/1 000, < 1/100); raro (≥ 1/10 000, < 1/1 000); molto raro (< 1/10 000) e non nota (la frequenza non può essere definita sulla base dei dati disponibili).
Il tempo mediano di trattamento con selpercatinib è stato di 30,09 mesi (LIBRETTO-001), 16,7 mesi (LIBRETTO-431) e 14,9 mesi (LIBRETTO-531).
Tabella 5 Reazioni averse al farmaco in pazienti che hanno ricevuto selpercatinib (N = 1188)


a Le infezioni delle vie urinarie comprendono infezione delle vie urinarie, cistite, urosepsi, infezione delle vie urinarie da Escherichia, pielonefrite da Escherichia, infezione renale,nitriti urinari presenti, pielonefrite, uretrite, infezione batterica delle vie urinarie e infezione urogenitale fungina.
b L’infezione polmonare comprende polmonite, infezione polmonare, infezione polmonare da aspirazione, empiema, consolidamento polmonare, infezione pleurica, polmonite batterica, polmonite da stafilococco, polmonite atipica, ascesso polmonare, polmonite da pneumocystis jirovecii, polmonite pneumococcica e polmonite respiratoria sinciziale virale, versamento pleurico infettivo e polmonite virale.
c Reazioni di ipersensibilità sono state caratterizzate da eruzione maculo-papulare, spesso preceduta da febbre associata a artralgia/mialgia durante il primo ciclo di trattamento del paziente (tipicamente tra i giorni 7-21).
d Ipersensibilità include ipersensibilità al farmaco e ipersensibilità.
e Cefalea include cefalea, cefalea sinusale e cefalea tensiva.
f Capogiri include capogiri, vertigine, presincope e capogiri posturali.
g QT prolungato all’elettrocardiogramma include QT prolungato all’elettrocardiogramma e intervallo QT anormale all’elettrocardiogramma.
h Ipertensione include ipertensione e pressione arteriosa aumentata.
i Emorragia include epistassi, emottisi, contusione, ematuria, emorragia rettale, emorragia vaginale, emorragia cerebrale, ematoma traumatico, sangue presente nell’urina, emorragia della congiuntiva, ecchimosi, sanguinamento gengivale, ematochezia, petecchie, vescicola ematica, ematoma spontaneo, ematoma della parete addominale, emorragia anale, angina bollosa emorragica, coaugulazione intravascolare disseminata, emorragia dell’occhio, emorragia gastrica, emorragia gastrointestinale, emorragia intracranica, emorragia sottocutanea, emorragia emorroidale, ematoma epatico, emorragia intra-addominale, emorragia della bocca, emorragia esofagea, ematoma pelvico, ematoma periorbitale, emorragia periorbitale, emorragia faringea, contusione polmonare, porpora, ematoma retroperitoneale, emorragia della pelle, emorragia subaracnoidea, diverticolo intestinale emorragico, ematoma dell’occhio, ematemesi, emorragia, ictus emorragico, emorragia epatica, emorragia laringea, emorragia del tratto gastrointestinale inferiore, melena, menorragia, sangue occulto positivo, emorragia post procedurale, emorragia post menopausale, emorragia della retina, emorragia della sclera, emorragia subdurale, emotorace traumatico, emorragia tumorale, emorragia del tratto gastrointestinale superiore, emorragia uterina ematoma al sito di iniezione del vaso, emartrosi ed ematoma.
j Malattia polmonare interstiziale/polmonite include malattia polmonare interstiziale, polmonite, polmonite da radiazione, malattia polmonare restrittiva, sindrome da distress respiratorio acuto, alveolite, bronchiolite, istiocitosi a cellule di Langerhans lesione polmonare da radiazione, malattia cistica polmonare, infiltrazione polmonare e opacità polmonare.
k Diarrea include diarrea, incontinenza anale, urgenza di defecazione, movimenti intestinali frequenti e ipermotilità gastrointestinale.
l Bocca secca include bocca secca e secchezza delle mucose.
m Dolore addominale include dolore addominale, dolore addominale superiore, fastidio addominale, dolore addominale inferiore e dolore gastrointestinale.
n Vomito include vomito, conati di vomito e rigurgito.
o Stomatite include stomatite, ulcerazione della bocca, infiammazione della mucosa, ed eruzione vescicolare della mucosa orale.
p Ascite chilosa include ascite chilosa e ascite chilosa (MedDRA LLTs).
q Eruzione cutanea include eruzione cutanea, eruzione maculo-papulare, dermatite, esfoliazione cutanea, eruzione maculare, eruzione eritomatosa, orticaria, dermatite allergica, eruzione esfoliativa, eruzione cutanea papulare, eruzione morbilliforme, eruzione pruriginosa, eruzione cutanea vescicolare, eruzione a farfalla, eruzione follicolare, eruzione cutanea generalizzata esantema pustoloso e reazione cutanea.
r Da dati di post-marketing
s L'epifisiolisi della testa del femore è stata comunemente osservata (6,4%) in pazienti pediatrici (<18 anni di età)
trattati con selpercatinib (n=47).
t La disfunzione erettile è stata osservata molto comunemente (12,4%) in pazienti di sesso maschile trattati con
selpercatinib negli studi clinici (n=986).
u Edema include edema periferico, edema facciale, edema periorbitale, gonfiore del viso, edema localizzato, gonfiore periferico, edema generalizzato, edema delle palpebre, gonfiore oculare, linfoedema, edema genitale, edema scrotale, angioedema, edema oculare, edema,edema dello scroto, edema cutaneo, gonfiore, edema orbitale, edema del testicolo e tumefazione vulvovaginale, tumefazione orbitale, edema del pene, gonfiore periorbitale e tumefazione della palpebra.
v Stanchezza include stanchezza, astenia e malessere.
w Sulla base delle valutazioni di laboratorio. La percentuale è calcolata in base al numero di pazienti con valutazione basale e almeno una valutazione post basale come denominatore.
Descrizione di reazioni averse selezionate in pazienti trattati con selpercatinib
Innalzamento delle aminotransferasi (AST / ALT aumentate)
Sulla base della valutazione di laboratorio, sono stati segnalati innalzamenti di ALT e AST rispettivamente nel 59,4 % e 61 % dei pazienti. Innalzamenti di grado 3 o 4 di ALT o AST sono stati segnalati rispettivamente nel 14,1% e 9,5% dei pazienti.
Il tempo mediano alla prima insorgenza è stato di 4,7 settimane per l’aumento di AST (intervallo: 0,7; 227,9) e di 4,4 settimane per l’aumento di ALT (intervallo: 0,9; 186,1) nello studio LIBRETTO-001; 5,1 settimane (intervallo: 0,7; 88,1) per l’aumento di AST e 5,1 settimane (intervallo: 0,7; 110,9) per l’aumento di ALT nello studio LIBRETTO-431; 6,1 settimane (intervallo: 0,1; 85,1) per l’aumento di AST e di 6,1 settimane (intervallo 0,1; 85,1) per l’aumento di ALT nello studio LIBRETTO-531.
Si raccomanda la modifica della dose per pazienti che sviluppano aumento delle ALT o AST di grado 3
o 4 (vedere paragrafo 4.2).
Prolungamento dell’intervallo QT
Nei 837 pazienti che hanno effettuato l’ECG nello studio LIBRETTO-001, la revisione dei dati
dell’ECG ha mostrato che l’8,1% dei pazienti aveva registrato un valore massimo post-basale di QTcF > 500 ms e il 21,6% dei pazienti un incremento massimo dal basale nell’intervallo QTcF > 60 ms.
Nei 156 pazienti dello studio LIBRETTO-431 ai quali è stato effettuato l’ECG, il 5,1% dei pazienti aveva un valore massimo di QTcF post-basale >500 msec e il 16,7% dei pazienti aveva un aumento massimo >60 msec rispetto al basale negli intervalli QTcF. Nei 191 pazienti dello studio LIBRETTO-531 ai quali è stato effettuato l’ECG, il 3,7% dei pazienti aveva un valore massimo di QTcF post-basale > 500 msec e il 17,8% dei pazienti aveva un innalzamento massimo di QTcF > 60 msec rispetto al basale.
Negli studi LIBRETTO-001, LIBRETTO-431 e LIBRETTO-531 non sono stati segnalati casi di torsioni di punta, eventi di grado ≥3 o aritmie clinicamente significative emergenti dal trattamento, tachicardia ventricolare, fibrillazione ventricolare o flutter ventricolare. Eventi fatali di morte improvvisa e arresto cardiaco sono stati riportati in pazienti con anamnesi cardiaca significativa. Considerando tutti gli studi, due pazienti (0,2%) hanno interrotto il trattamento con selpercatinib a causa del prolungamento dell'intervallo QT.
Retsevmo può richiedere interruzione o modifica della dose (vedere paragrafi 4.2 e 4.4).
Ipertensione
Negli 837 pazienti nei quali è stata misurata la pressione arteriosa nello studio LIBRETTO-001, l’aumento mediano massimo della pressione sistolica dal basale era di 32 mmHg (intervallo: -15, +100). I risultati sulla pressione diastolica erano simili ma gli incrementi sono stati di minore entità.
Nello studio LIBRETTO-001, solo il 10,3% dei pazienti ha mantenuto il livello basale durante il trattamento; il 40,7% ha avuto un incremento di 1 grado, il 38,5% di 2 gradi e il 9,8% di 3 gradi. Un evento avverso di ipertensione mergente dal trattamento è stato segnalato nel 44,8% dei pazienti con storia di ipertensione (28,2% di grado 3, 4) e nel 41,7% dei pazienti senza storia di ipertensione (14,1% di grado 3, 4).
Nei 154 pazienti trattati con selpercatinib che hanno avuto misurazioni della pressione arteriosa nello studio LIBRETTO-431, il 23,4% dei pazienti trattati con selpercatinib ha mantenuto il grado basale durante il trattamento, il 49,4% ha avuto un incremento di 1 grado, il 22,7% ha avuto un incremento di 2 gradi e il 3,3% ha avuto un incremento di 3 gradi.
Nei 192 pazienti trattati con selpercatinib che hanno avuto misurazioni della pressione arteriosa nello studio LIBRETTO-531, il 20,8% dei pazienti trattati con selpercatinib ha mantenuto il grado basale durante il trattamento, il 43,8% ha avuto un incremento di 1 grado, il 27,6% ha avuto un incremento di 2 gradi e il 6,8% ha avuto un incremento di 3 gradi.
Complessivamente, un totale pari al 19,8% dei pazienti nello studio LIBRETTO-001, 20,3% dei pazienti nello studio LIBRETTO-431 e 19,2% di pazienti nello studio LIBRETTO-531 ha manifestato ipertensione di grado 3 emergente dal trattamento (definita come una pressione sistolica massima superiore a 160 mmHg). Ipertensione di grado 4 emergente dal trattamento è stata riportata nello0,1% dei pazienti nello studio LIBRETTO-001 e nessuna è stata riportata negli studi LIBRETTO-431 e LIBRETTO-531.
Due pazienti (0,2%) hanno interrotto permanentemente il trattamento a causa dell’ipertensione nello studio LIBRETTO-001 e nessun paziente negli studi LIBRETTO-431 e LIBRETTO-531. È raccomandata la modifica della dose in pazienti che sviluppano ipertensione (vedere paragrafo 4.2).
Selpercatinib deve essere interrotto in maniera definitiva se l’ipertensione clinicamente significativa non può essere controllata con terapia anti-ipertensiva (vedere paragrafo 4.4).
Ipersensibilità
Segni e sintomi di ipersensibilità hanno incluso febbre, eruzione cutanea e artralgia o mialgia con concomitante diminuzione delle piastrine o incremento dell’aminotransferasi.
Nello studio LIBRETTO-001, il 24,0% (201/837) dei pazienti trattati con selpercatinib aveva ricevuto precedentemente immunoterapia con anti PD-1/PD-L1. L’ipersensibilità si è verificata in un totale del 5,7% (48/837) dei pazienti che hanno ricevuto selpercatinib, inclusa l’ipersensibilità di grado 3 nell’1,9% (16/837) dei pazienti.
Dei 48 pazienti con ipersensibilità nello studio LIBRETTO-001, il 54,2% (26/48) era affetto da NSCLC e aveva ricevuto precedente immunoterapia con anti-PD-1/PD-L1.
Nello studio LIBRETTO-001, ipersensibilità di grado 3 si è manifestata nel 3,5% (7/201) dei pazienti precedentemente trattati con immunoterapia con anti-PD-1/PD-L1.
Nello studio LIBRETTO-001 il tempo mediano per l’insorgenza era di 1,9 settimane (intervallo tra 0,7 settimane e 203,9 settimane): 1,7 settimane nei pazienti con precedente immunoterapia anti-PD/PD-L1 e 4,4 settimane nei pazienti che erano naïve all’immunoterapia anti-PD-1/PD-L1.
Lo studio LIBRETTO-431 ha arruolato pazienti con NSCLC avanzato o metastatico. L'ipersensibilità si è verificata in un totale dell’1,9% (3/158) dei pazienti trattati con selpercatinib, inclusa ipersensibilità di grado 3 nello 0,6% (1/158) dei pazienti. In un'analisi integrata dei pazienti con NSCLC che hanno ricevuto selpercatinib e che erano stati precedentemente trattati con terapia anti-PD-1/PD-L1 sulla base degli studi LIBRETTO-001 e LIBRETTO-431 (N=205), l'ipersensibilità si è verificata nel 16,6% dei pazienti, inclusa l'ipersensibilità ≥Grado 3 nel 5,9% dei pazienti.
Lo studio LIBRETTO-531 ha arruolato pazienti con MTC avanzato o metastatico. L'ipersensibilità si è verificata in 1 paziente (0,5% [1/193]) in trattamento con selpercatinib. Questo paziente ha manifestato ipersensibilità di grado 3.
Retsevmo potrebbe richiedere interruzione o modifiche della dose (vedere paragrafo 4.2).
Emorragie
Eventi emorragici di Grado ≥ 3 si sono verificati nel 2,5% dei pazienti trattati con selpercatinib negli studi LIBRETTO-001, LIBRETTO-431 e LIBRETTO-531. Lo studio LIBRETTO-001 includeva 4 pazienti (0,5%) con eventi emorragici fatali, due casi di emorragia cerebrale e un caso ciascuno di emorragia dal sito di tracheostomia e di emottisi. Negli studi LIBRETTO-431 e LIBRETTO-531 non sono stati riportati eventi emorragici fatali nei pazienti trattati con selpercatinib. Il tempo mediano all’insorgenza è stato di 34,1 settimane (intervallo: tra 0,1 e 234,6 settimane) nello studio LIBRETTO-001, 16,8 settimane (intervallo tra 1,1 e 94,1 settimane) nello studio LIBRETTO-431 e 10,7 settimane (intervallo tra 1,0 e 124,1 settimane) nello studio LIBRETTO-531.
Selpercatinib deve essere interrotto definitivamente nei pazienti con emorragia potenzialmente letale o
ricorrente severa (vedere paragrafo 4.2).
Informazioni aggiuntive su popolazioni particolari
Pazienti pediatrici
Nello studio LIBRETTO-001 vi erano 3 pazienti di età < 18 anni (intervallo: 15-17 anni) con MTC con mutazione RET. Nello studio LIBRETTO-121 vi erano 11 pazienti di età < 18 anni (intervallo: 2-17 anni) con MTC con mutazione di RET, 13 pazienti di età < 18 anni (intervallo 6–17) con carcinoma tiroideo positivo per fusione di RET, e 6 pazienti di età < 18 anni (intervallo 5–15) con tumori solidi con alterazioni di RET, di cui 3 erano pazienti con tumori solidi positivi per fusione di RET. Nello studio LIBRETTO-531 c’era 1 paziente di 12 anni di età con MTC con mutazione RET.
Casi di epifisiolisi della testa del femore sono stati riportati in pazienti < 18 anni di età trattati con selpercatinib (vedere paragrafo 4.4). Non sono stati identificati altri risultati sulla sicurezza in ragazzi di età inferiore a 18 anni.
Tabella delle reazioni avverse al farmaco nello studio LIBRETTO-121
La frequenza e la gravità delle ADR segnalate in pazienti di età ≤ 21 anni trattati con selpercatinib nello Studio LIBRETTO-121 sono mostrate nella Tabella 6.
Le ADR sono classificate in accordo alla classificazione per sistemi e organi secondo MedDRA e per frequenza.
I gruppi di frequenza sono definiti secondo la seguente convenzione: molto comune (≥ 1/10); comune (≥ 1/100, < 1/10); non comune (≥ 1/1 000, < 1/100); raro (≥ 1/10 000, < 1/1 000); molto raro (< 1/10 000) e non nota (la frequenza non può essere definita sulla base dei dati disponibili).
Il tempo mediano di trattamento con selpercatinib è stato di 30,26 mesi.
Tabella 6 Reazioni avverse al farmaco nei pazienti trattati con selpercatinib nello Studio LIBRETTO-121 (n = 36)


# Tra i 36 pazienti nello studio LIBRETTO-121, 31 pazienti avevano meno di 18 anni e 5 pazienti avevano un’età compresa tra 18 e 21 anni.
a Le infezioni delle vie urinarie includono infezioni delle vie urinarie e cistite.
b La polmonite include polmonite e polmonite da virus respiratorio sinciziale.
c Le reazioni di ipersensibilità sono state caratterizzate da un’eruzione maculopapulare spesso preceduta da febbre con artralgie/mialgie associate durante il primo ciclo di trattamento del paziente (in genere tra i Giorni 7-
21).
d L’ipersensibilità include l’ipersensibilità al farmaco e l’ipersensibilità.
e L’ipotiroidismo include ipotiroidismo, aumento dell’ormone tireostimolante nel sangue e aumento della tireoglobulina.
f Il prolungamento dell’intervallo QT all’elettrocardiogramma include il prolungamento del QT all’elettrocardiogramma e il prolungamento del complesso QRS all’elettrocardiogramma.
g L’emorragia include epistassi, ematuria, tempo di tromboplastina parziale attivata prolungato, emorragia anale,
presenza di sangue nelle urine, emottisi, menorragia ed emorragia orale.
h La diarrea include diarrea e incontinenza anale.
i Il dolore addominale include dolore addominale, dolore addominale superiore e fastidio addominale.
j L’eruzione cutanea include eruzione cutanea maculo-papulare, eruzione cutanea, eruzione cutanea eritematosa e
orticaria.
k Il dolore muscoloscheletrico include artralgia, dolore agli arti, dolore dorsale, dolore toracico non cardiaco,
dolore osseo, dolore toracico muscoloscheletrico, dolore muscoloscheletrico e dolore al collo.
l Disfunzione erettile nei pazienti di sesso maschile trattati con selpercatinib. Denominatore corretto a causa di un evento specifico per genere per i maschi (n = 19).
m L’affaticamento include affaticamento, astenia e malessere.
n L’edema comprende edema facciale, edema periferico, edema periorbitale, edema generalizzato, edema localizzato e gonfiore.
o Sulla base delle valutazioni di laboratorio. La percentuale è calcolata in base al numero di pazienti conbvalutazione al basale e almeno una valutazione post-basale come denominatore.
Anziani
Nei pazienti che hanno ricevuto selpercatinib nello studio LIBRETTO-001, il 24,7% era di età compresa tra 65 e 74 anni, 8,6% era di età compresa tra 75 e 84 anni e l’1,0% di età ≥ 85 anni.
Nello studio LIBRETTO-431, il 26,6% dei pazienti che hanno ricevuto selpercatinib aveva un'età compresa tra 65 e 74 anni, il 9,5% aveva un'età compresa tra 75 e 84 anni e l'1,3% aveva un'età ≥85 anni. Nello studio LIBRETTO-531, il 20,2% dei pazienti che hanno ricevuto selpercatinib aveva un'età compresa tra 65 e 74 anni, il 5,2% aveva un’età compresa tra 75 e 84 anni e nessuno aveva ≥85 anni. La frequenza degli eventi avversi seri è stata più alta nei pazienti di età compresa tra 65 e 74 anni (58,0%), 75-84 anni (62,5%) e ≥ 85 anni (100,0%) rispetto ai pazienti di età < 65 anni (46,7%) nello studio LIBRETTO-001 e nello studio LIBRETTO-431, ≥65-74 anni (38,1%), 75-84 anni (46,7%), ≥85 anni (50,0%), rispetto a quelli di età < 65 anni (31,3%). Nello studio LIBRETTO-531 la frequenza degli eventi avversi seri è stata più alta nei pazienti di età compresa tra 75 e 84 anni (50%) rispetto ai pazienti di età <65 anni (20.8%) e di età compresa tra 65-74 anni (17.9%).
Nello studio LIBRETTO-001 la frequenza degli eventi avversi (EA) che hanno portato all’interruzione di selpercatinib è stata più alta nei pazienti di età compresa tra 65 e 74 anni (10,1%), 75-84 anni (19,4%) e ≥ 85 anni (37,5%) rispetto ai pazienti di età < 65 anni (7,6%). Nello studio LIBRETTO-431, la frequenza di eventi avversi che hanno portato all'interruzione del trattamento con selpercatinib è stata più alta nei pazienti di età compresa tra 65 e 74 anni (14,3%), 75-84 anni (20,0%) rispetto ai pazienti <65 anni (7,1%) di età. Nessun paziente di età ≥85 anni ha interrotto il trattamento con selpercatinib a causa di eventi avversi.
Nello studio LIBRETTO-531, la frequenza degli eventi avversi che hanno portato all'interruzione di selpercatinib è stata più alta nei pazienti di età compresa tra 75 e 84 anni (10%) e ≥65-74 anni (7,7%) rispetto ai pazienti di <65 anni (3,5%).
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l’autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite l’Agenzia Italiana del Farmaco Sito web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
4.9 Sovradosaggio
Non sono stati stabiliti i sintomi del sovradosaggio. In caso di sospetto sovradosaggio, deve essere fornita una terapia di support.
5.1 Proprietà farmacodinamiche
Categoria farmacoterapeutica: antineoplastici e agenti immunomodulatori, agenti antineoplastici inibitori delle protein chinasi, codice ATC: L01EX22.
Meccanismo d’azione
Selpercatinib è un inibitore del recettore tirosin chinasico riarrangiato durante la transfezione (rearranged during transfection, RET). Selpercatinib ha inibito isoforme wild type di RET e numerose isoforme mutate di RET così come VEGFR1 e VEGFR3 (vascular endothelial growth factor receptor)con valori di IC50 che vanno da 0,92 nM a 67,8 nM. In altri saggi enzimatici, selpercatinib ha inibito anche FGFR 1, 2 e 3 (fibroblast growth factor receptor) a concentrazioni più alte di quelle che erano ancora clinicamente ottenibili. In un saggio di legame, alla concentrazione di 1 µM di selpercatinib, è stata osservata una significativa attività di legame antagonista (> 50%) per il trasportatore 5-HT (serotonina) (70,2% antagonista) e per l’adrenorecettore α2C (51,7% antagonista). La concentrazione di 1 µM è circa 7 volte superiore alla concentrazione massima plasmatica libera della dose efficace di selpercatinib.
Alcune mutazioni puntiformi di RET o riarrangiamenti cromosomici che coinvolgono fusioni in frame di RET con vari partner possono dare origine a proteine RET di fusione chimeriche costitutivamente attive, che possono agire come driver oncogenici, promuovendo la proliferazione di linee cellulari tumorali. Nei modelli tumorali in vitro e in vivo selpercatinib ha dimostrato attività antitumorale in cellule portatrici dell’attivazione costitutiva della proteina RET risultante da fusioni e mutazioni geniche, inclusi CCDC6-RET, KIF5B-RET, RET V804M e RET M918T. In aggiunta, selpercatinib ha mostrato attività antitumorale nei topi in cui era stato impiantato a livello intracranico un tumore RET fusione-positivo derivato da un paziente.
Effetti farmacodinamici
Elettrofisiologia cardiaca
In uno studio approfondito sul QT con 32 soggetti sani come controllo positivo, non sono stati rilevati cambiamenti considerevoli (cioè > 20 ms) nell’intervallo QTcF a concentrazioni di selpercatinib simili a quelle osservate con le schede di dosaggio terapeutico. Un’analisi esposizione-risposta ha indicato che concentrazioni al di sopra di quelle terapeutiche possono portare ad un incremento nel QTc > 20 ms.
Nei pazienti che hanno ricevuto selpercatinib, è stato segnalato un prolungamento dell’intervallo QT.
Di conseguenza, nei pazienti può essere necessaria interruzione o modifica della dose (vedere
paragrafi 4.2 e 4.4).
Efficacia e sicurezza clinica
L’efficacia di Retsevmo è stata valutata in pazienti adulti con NSCLC avanzato RET fusione-positivo, cancro della tiroide RET fusione-positivo, altri tumori solidi RET fusione-positivi e in pazienti adulti e adolescenti con MTC e mutazione di RET arruolati in uno studio multicentrico di fase 1/2, in aperto, a braccio singolo: lo studio LIBRETTO-001. L'efficacia di Retsevmo nel NSCLC RET fusione positivo è stata confermata nello studio di fase 3 LIBRETTO-431 (vedere paragrafo NSCLC RET fusione positivo naïve al trattamento). L'efficacia di Retsevmo nel MTC con mutazione di RET, è stata confermata nello studio di fase 3 LIBRETTO-531 (vedere paragrafo Cancro midollare della tiroide (MTC) con mutazione di RET naïve al trattamento con vandetanib e cabozantinib).
Lo studio LIBRETTO-001 era costituito da due parti: la fase 1 (incremento della dose) e la fase 2 (espansione della dose). L’obiettivo primario della fase 1 è stato determinare la dose di selpercatinib raccomandata per la fase 2. L’obiettivo primario della fase 2 è stato valutare l’attività anti-tumorale di selpercatinib attraverso la determinazione del tasso di risposta obiettiva (objective response rate, ORR), valutato da un comitato di revisione indipendente. Sono stati arruolati pazienti con patologia misurabile o non-misurabile come determinato dai criteri RECIST 1.1, con evidenza di un’alterazione del gene RET nel tumore. Pazienti con metastasi al sistema nervoso centrale (SNC) erano eleggibili se stabili, mentre sono stati esclusi pazienti con tumore primario sintomatico a carico del SNC e metastasi sintomatiche, carcinomatosi leptomeningea o compressione del midollo spinale. Sono stati esclusi pazienti con alterazione driver primaria nota diversa da RET, patologia cardiovascolare in atto
clinicamente significativa o anamnesi di infarto del miocardio, intervallo QTcF > 470 ms.
Pazienti nella fase 2 dello studio hanno ricevuto Retsevmo 160 mg per via orale due volte al giorno fino a tossicità inaccettabile o progressione di malattia. L’identificazione di un’alterazione del gene RET è stata determinata in maniera prospettica in laboratori locali che usavano sequenziamento di nuova generazione (next generation sequencing, NGS), reazioni a catena della polimerasi (polymerase chain reaction, PCR) o ibridizzazione fluorescente in situ (fluorescence in situ hybridization, FISH).
La principale misura dell’esito di efficacia è stata l’ORR in accordo al RECIST v1.1 come valutato dal Comitato di Revisione Indipendente (Independent Review Committee, IRC). Gli esiti di efficacia secondari hanno incluso la durata della risposta (DOR), la progressione libera da malattia (progression free survival, PFS) e la sopravvivenza complessiva (overall survival, OS).
NSCLC RET fusione-positivo naïve al trattamento
LIBRETTO-431
L'efficacia di Retsevmo nel NSCLC RET fusione positivo è stata confermata nello studio LIBRETTO-431, uno studio di confronto multicentrico, randomizzato, in aperto, di fase 3, che ha confrontato selpercatinib con la terapia a base di platino e pemetrexed con o senza pembrolizumab in pazienti con NSCLC avanzato o metastatico RET fusione positivo. Erano eleggibili i pazienti adulti con NSCLC istologicamente confermato, non resecabile, localmente avanzato o metastatico senza precedente terapia sistemica per la malattia metastatica. Erano eleggibili anche i pazienti che avevano ricevuto una terapia adiuvante o neoadiuvante se l'ultima dose di trattamento sistemico era stata completata almeno 6 mesi prima della randomizzazione. I pazienti hanno ricevuto 160 mg di selpercatinib due volte al giorno (dose iniziale) o una terapia a base di platino e pemetrexed con o senza pembrolizumab. I pazienti sono stati stratificati in base alla regione geografica (Asia orientale vs. altrove), allo stato rispetto alle metastasi cerebrali valutate dallo sperimentatore al basale (assenti o sconosciute vs presenti) e se lo sperimentatore aveva intenzione (prima della randomizzazione) di trattare il paziente con o senza pembrolizumab. La misura primaria dell'outcome di efficacia era la PFS secondo RECIST 1.1 mediante revisione centralizzata indipendente in cieco (BICR). Gli esiti secondari di efficacia includevano OS, ORR/DOR/tasso di controllo della malattia (Disease Control Rate,DCR) mediante BICR, ORR/DOR intracranico mediante BICR e tempo di deterioramento dei sintomi polmonari mediante NSCLC-questionario di valutazione dei sintomi (Symptom Assessment Questionnaire,SAQ).
Dei 261 pazienti arruolati e randomizzati nella popolazione intention-to treat (ITT) dello studio LIBRETTO-431, 212 sono stati stratificati in base al fatto che lo sperimentatore intendesse che il paziente ricevesse pembrolizumab (prima della randomizzazione), per formare la popolazione ITT-Pembrolizumab. Nella popolazione ITT-Pembrolizumab, 129 pazienti hanno ricevuto selpercatinib mentre 83 hanno ricevuto chemioterapia a base di pembrolizumab con pemetrexed. L'età mediana dei pazienti nella popolazione ITT-Pembrolizumab era di 61,5 anni (intervallo da 31 a 84 anni). Il 53,3% dei pazienti era di
sesso femminile. Il 41,3% dei pazienti erano bianchi, il 56,3% asiatici, l'1% neri. Il 67,9% non aveva mai fumato. Nella popolazione ITT Pembrolizumab, il 93% presentava una malattia metastatica e il 20,3% dei pazienti presentava metastasi del SNC al basale. Il performance status ECOG è stato riportato come 0-1 (96,7%) o 2 (3,3%). Il partner di fusione più comune è stato KIF5B (44,8%), seguito da CCDC6 (9,9%). Lo studio ha raggiunto il suo endpoint primario di miglioramento della PFS in entrambe le popolazioni ITT-Pembrolizumab e ITT. I risultati primari di efficacia per la popolazione di ITT-Pembrolizumab per i pazienti naïve al trattamento con NSCLC RET fusione positivo sono riassunti nella Tabella 7 e nella Figura 1.
Tabella 7 LIBRETTO-431: Sintesi dei dati di efficacia (valutazione BICR, popolazione ITT-Pembrolizumab)

IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile, RP = risposta parziale
*La durata mediana del follow-up è stata di 17,97 mesi (25°, 75° percentile: 12,32; 21,03) nel braccio
selpercatinib e di 14,55 mesi (25°, 75° percentile: 9,69; 20,73) nel braccio di controllo.
Data di cut off dei dati: 01 maggio 2023.
Figura 1. LIBRETTO-431: Diagramma di Kaplan-Meier della sopravvivenza libera da progressione (valutazione BICR, popolazione ITT-Pembrolizumab)

Data di cut-off dei dati: 01 maggio 2023
La OS non era matura al momento dell'analisi primaria della PFS.
Al momento di un’analisi descrittiva ad interim aggiornata dell’OS (43% degli eventi di OS prespecificati necessari per l’analisi finale, con data lock dei dati del 1°maggio 2024), nella popolazione ITT, sono stati osservati 75 eventi nei due bracci e l’Hazard Ratio (HR) è stato di 1,259 [(IC 95%: 0,777, 2,040); p= 0, 3496]. A 30 mesi, la sopravvivenza globale stimata è stata del 71% (IC 95%: 63,78) e del 76% (IC 95%: 66,84) rispettivamente nel braccio selpercatinib e nel braccio di controllo. La OS può essere influenzata dallo squilibrio nelle terapie post-progressione. Dei 68 pazienti del braccio di controllo che hanno avuto progressione della malattia, 50 pazienti (74%) hanno ricevuto selpercatinib alla progressione. Dei 71 pazienti del braccio selpercatinib che hanno avuto progressione della malattia, 16 (23%) hanno ricevuto chemioterapia e/o terapia con inibitori del checkpoint immunitario e 44 (62%) hanno continuato a ricevere selpercatinib.
Nella popolazione ITT-Pembrolizumab, selpercatinib ha ritardato significativamente il tempo di peggioramento dei sintomi NSCLC riferiti dai pazienti, come misurato dal punteggio totale del NSCLC-SAQ (aumento di ≥ 2 punti) rispetto al controllo (HR: 0,34 [IC 95%: 0,20; 0,55]; il tempo mediano non è stato raggiunto per il braccio selpercatinib rispetto a 1,9 mesi [IC 95%: 0,7; 6,6]) per il braccio di controllo. Inoltre, selpercatinib ha ritardato significativamente il tempo necessario al deterioramento confermato della funzione fisica e ha mantenuto la qualità complessiva della vita nel tempo.
LIBRETTO-001
Dei 362 pazienti con NSCLC RET fusione positivo arruolati nello studio LIBRETTO-001, 69 erano naïve al trattamento. L’età mediana era 63 anni (intervallo da 23 a 92 anni). Il 62,3% dei pazienti erano di sesso femminile, il 69,6% bianchi, il 18,8% asiatici, il 5,8% neri e il 69,6% non aveva mai fumato. La maggior parte dei pazienti (98,6%), presentavano malattia metastatica all’arruolamento e il 23,2% aveva metastasi del SNC al basale, come valutato dallo sperimentatore. L’ECOG performance status è stato riportato come 0-1 (94,2%) o 2 (5,8%). Il partner di fusione più comune era il KIF5B (69,6%), seguito da CCDC6 (14,5%) e infine NCOA4 (1,4%). I risultati di efficacia per pazienti con NSCLC RET fusione positivo naïve al trattamento sono riassunti nella tabella 8.
Tabella 8 LIBRETTO-001: Risposta obiettiva e durata della risposta
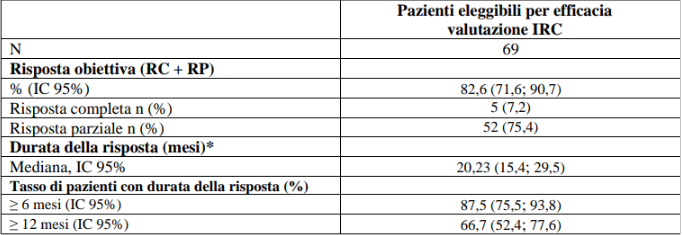
IC = Intervallo di confidenza, RC = risposta completa, RP = risposta parziale
*La durata mediana del follow up è stata 37,09 mesi (25°, 75° percentile: 24,0; 45,1)
Data di cut-off dei dati: 13 gennaio 2023
NSCLC RET fusione positivo precedentemente trattato
Nello studio LIBRETTO-001 un totale di 247 pazienti avevano ricevuto un precedente trattamento chemioterapico a base di platino. L’età mediana era 61 anni (intervallo da 23 a 81 anni). Il 56,7% dei pazienti erano di sesso femminile. La categorizzazione dei pazienti è stata: 43,7% bianchi, il 47,8% asiatici, il 4,9% neri e il 66,8% non aveva mai fumato. La maggior parte dei pazienti (98,8%) presentava malattia metastatica all’arruolamento e il 31,2% aveva metastasi del SNC al basale come valutato dallo sperimentatore. L’ECOG performance status è stato riportato come 0-1 (97,1%) o 2 (2,8%). Il partner di fusione più comune è stato KIF5B (61,9%), seguito da CCDC6 (21,5%) e infine NCOA4 (2,0%). La mediana del numero di precedenti terapie sistemiche è stata 2 (intervallo 1–15) e il 43,3% (n = 107/247) ha ricevuto 3 o più precedenti regimi sistemici; trattamenti precedenti includevano terapia anti PD1/PD-L1 (58,3%), inibitori multi-kinasi (multi-kinase inhibitor, MKI) (31,6%) e taxani (34,8%); il 41,3% aveva ricevuto altre terapie sistemiche. I risultati di efficacia per il precedente trattamento del NSCLC per i pazienti RET fusione positivi sono riassunti nella tabella 9.
Tabella 9 LIBRETTO-001: Risposta obiettiva e durata della risposta
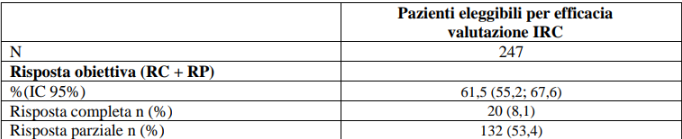
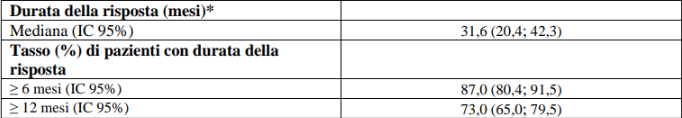
IC = Intervallo di confidenza, RC = risposta completa, RP = risposta parziale
*La durata mediana del follow-up è stata 39,52 mesi (25°, 75° percentile: 24,6; 45,0).
Data di cut-off dei dati: 13 gennaio 2023.
Risposta del SNC nel NSCLC RET fusione-positivo
Nello studio LIBRETTO-431 l'ORR del SNC valutato mediante BICR è stato dell'82,4% (14/17 IC
95%: 56,6; 96,2) nei 17 pazienti con metastasi cerebrali misurabili al basale trattati con selpercatinib, rispetto al 58,3% (7/12 IC 95%: da 27,7 a 84,4) nei 12 pazienti nel braccio di controllo della popolazione ITT-Pembrolizumab. La RC è stata osservata in 6/17 (35,3%) dei pazienti nel braccio selpercatinib rispetto a 2/12 (16,7%) pazienti nel braccio di controllo. Con un tempo di follow-up mediano per DOR di 9,92 mesi (IC 95%: 7,66; 18,10) nel braccio selpercatinib e di 12,68 mesi (IC 20 95%: 2,79; NS) nel braccio di controllo, la DOR mediana non è stata raggiunta per selpercatinib (IC 95%: 7,62; NS) rispetto a 13,4 mesi (IC 95%: 3,45; NS) del controllo. In 192 pazienti con scansioni intracraniche disponibili al basale, l'hazard ratio causa-specifica per il tempo alla progressione del SNC, come valutato mediante BICR, è stato di 0,28; IC 95%: 0,12; 0,68 (HR di 0,17; IC 95%: 0,04; 0,69 per 150 pazienti senza metastasi intracraniche basali e HR di 0,61; IC 95%: 0,19; 1,92 per 42 pazienti con metastasi intracraniche al basale). 8 pazienti (6,7%) nel braccio selpercatinib hanno avuto un primo evento di progressione del SNC rispetto a 13 pazienti (18,1%) nel braccio di controllo.
Nello studio LIBRETTO-001 l’ORR nel SNC valutata dall’IRC è stata 84,6% (22/26; IC 95%: 65,1;
95,6) nei 26 pazienti con malattia misurabile. L’RC è stata osservata in 7 pazienti (26,9%) e l’RP in 15 pazienti (57,5%). La DOR mediana del SNC è stata 9,36 mesi (IC 95%: 7,4; 15,3).
Cancro della tiroide RET fusione positivo naïve al trattamento sistemico
Dei pazienti con cancro della tiroide RET fusione positivo naïve alla terapia sistemica diversa dallo iodio radioattivo e arruolati nello studio LIBRETTO-001, 24 pazienti hanno avuto l'opportunità di essere seguiti per almeno 6 mesi e sono stati considerati eleggibili per la valutazione dell’efficacia.
L'età mediana era di 60,5 anni (intervallo (range) da 20 a 84 anni). Il 58,3% dei pazienti era di sesso maschile. Il 75% dei pazienti erano bianchi. L’ECOG performance status è stato riportato come 0-1 (95,8%) o 2 (4,2%). Il 100% dei pazienti aveva una storia di malattia metastatica. 22 dei 24 pazienti (91,7%) hanno ricevuto iodio radioattivo prima dell'arruolamento e quindi sono stati considerati refrattari allo iodio radioattivo. Le diverse istologie rappresentate nei 24 pazienti includevano: papillare (n=23) e scarsamente differenziato (n=1). Il partner di fusione più comune è stato CCDC6 (62,5%) seguito da NCOA4 (29,2%). I risultati di efficacia per i pazienti con cancro della tiroide RET fusione positivo naïve al trattamento sistemico, sono riassunti nella Tabella 10.
Tabella 10 LIBRETTO-001: Risposta obiettiva e durata della risposta
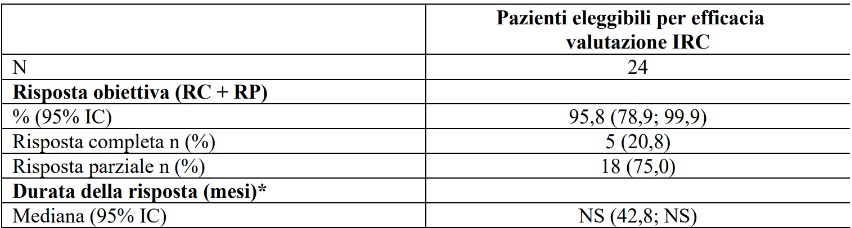
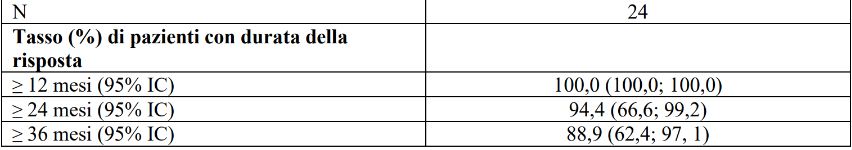
IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile RP = risposta parziale
* La durata mediana del follow-up è stata 54,80 mesi (25°, 75° percentile: 32,3; 62,5)
Data di cut-off dei dati: 14 febbraio 2025
Cancro della tiroide RET fusione-positivo pretrattato
Dei pazienti con cancro della tiroide RET fusione-positivo precedentemente trattati con terapia sistemica diversa dallo iodio radioattivo e che sono stati arruolati nello studio LIBRETTO-001, 41 pazienti hanno avuto la possibilità di essere seguiti per almeno 6 mesi e sono stati considerati eleggibili per la valutazione dell’efficacia. L’età mediana è stata di 58 anni (intervallo tra 25 e 88 anni), il 43,9% dei pazienti era di sesso maschile, il 58,5% bianchi, mentre il 29,3% asiatici, il 7,3% neri. L’ECOG performance status era 0-1 (92,7%) o 2 (7,3%). Il 100% dei pazienti aveva malattia metastatica. I pazienti avevano ricevuto una mediana di 3 terapie sistemiche precedenti (intervallo: 1-7). Le terapie precedenti più comuni includevano iodio radioattivo (73,2%), MKI (85,4%). Il 9,8% aveva ricevuto altre terapie sistemiche. Le diverse istologie rappresentate tra i 41 pazienti includevano: papillare (n = 31), scarsamente differenziato (n = 5), anaplastico (n = 4) e a cellule di Hurtle (n = 1). Il partner di fusione più comune è stato CCDC6 (61,0%) seguito da NCOA4 (19,5%).
I risultati di efficacia nel cancro della tiroide precedentemente trattato RET fusione-positivo sono riassunti nella tabella 11.
Tabella 11 LIBRETTO-001 Risposta obiettiva e durata della risposta

IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile, RP = risposta parziale
*La mediana della durata del follow up è stata 33,87 mesi (25°, 75° percentile: 12,9; 44,8)
Data di cut-off dei dati: 13 gennaio 2023.
Cancro midollare della tiroide (MTC) con mutazione di RET naïve al trattamento con vandetanib e cabozantinib
LIBRETTO-531
L'efficacia di Retsevmo nel MTC con mutazione di RET, è stata confermata nello studio LIBRETTO-531, uno studio comparativo multicentrico di fase 3, randomizzato, in aperto, che ha confrontato selpercatinib con cabozantinib o vandetanib a scelta del medico, in pazienti con MTC in progressione, avanzato, naïve agli inibitori delle chinasi e con mutazione di RET. Erano idonei i pazienti adulti o adolescenti con MTC istologicamente confermato, non resecabile, localmente avanzato o metastatico senza precedente trattamento con un inibitore delle chinasi. I pazienti hanno ricevuto 160 mg di selpercatinib due volte al giorno (dose iniziale) o in base alla scelta del medico cabozantinib (140 mg una volta al giorno) o vandetanib (300 mg una volta al giorno). I pazienti sono stati stratificati in base alla mutazione di RET (M918T vs. altro) e al trattamento scelto, se randomizzati al braccio di controllo (cabozantinib vs vandetanib). L'outcome primario di efficacia era la PFS secondo RECIST 1.1 valutata dal BICR. I principali risultati secondari di efficacia includevano la sopravvivenza libera da fallimento del trattamento (treatment failure-free survival, TFFS) e la tollerabilità comparativa, mentre altri esiti secondari di efficacia includevano OS e ORR/DOR valutati dal BICR.
Dei 291 pazienti arruolati e randomizzati nello studio LIBRETTO-531 della popolazione ITT, 193 sono stati randomizzati al braccio selpercatinib e 98 sono stati randomizzati al braccio di controllo.
Dei 98 pazienti randomizzati al braccio di controllo, 73 sono stati stratificati a cabozantinib e 25 sono stati stratificati a vandetanib. L'età mediana dei pazienti nella popolazione ITT era di 55 anni (intervallo: da 12 a 84 anni). Il 37,1% dei pazienti era di sesso femminile. Il 69,4% dei pazienti erano bianchi, il 27,7% erano asiatici, il 2,9% erano neri. La maggior parte dei pazienti (77%) presentava una malattia metastatica al momento dell'arruolamento. L’ECOG performance status è stato riportato come 0-1 (98,3%) o 2 (1%). La mutazione più comune è stata l’M918T (62,5%). Lo studio ha raggiunto il suo endpoint primario di miglioramento della PFS nella popolazione ITT. I risultati di efficacia per la popolazione ITT sono riassunti nella Tabella 12 e nella Figura 2.
Tabella 12 LIBRETTO-531: riassunto dei dati di efficacia (valutazione BICR, popolazione ITT)
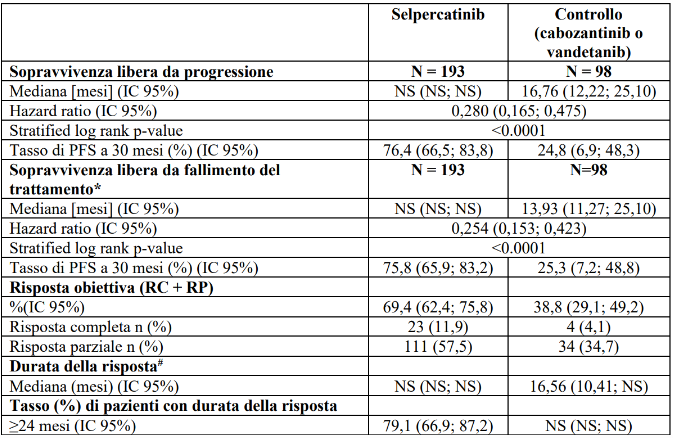
IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile, RP = risposta parziale
*La sopravvivenza libera da fallimento del trattamento è definita come il tempo che intercorre tra la randomizzazione e il primo verificarsi di: progressione radiografica documentata della malattia secondo RECIST 1.1, o tossicità inaccettabile che porta all'interruzione del trattamento come valutato dallo sperimentatore, o morte per qualsiasi causa.
#Durata mediana del follow-up è stata di 11,14 mesi (25° e 75° percentile: 5,62; 16,62) nel braccio selpercatinib e di 12,81 mesi (25° e 75° percentile: 6,34; 15,51) nel braccio di controllo.
Data di cut-off dei dati: 22 maggio 2023
Figura 2. LIBRETTO-531: Diagramma di Kaplan-Meier della sopravvivenza libera da
progressione (valutazione BICR, popolazione ITT)

Data di cut-off dei dati: 22 maggio 2023
Al momento dell'analisi primaria della PFS, sono stati osservati 18 eventi di OS nei due bracci. Nella popolazione ITT, l'OS HR era 0,374 ([IC 95%: 0,147; 0,949]). Il tasso di censura è stato del 95,9% nel braccio selpercatinib e dell'89,8% nel braccio di controllo.
La tollerabilità è stata valutata in 242 pazienti (braccio selpercatinib, N=161; braccio di controllo, N=81). Il braccio selpercatinib ha mostrato in maniera statisticamente significativa, una percentuale più bassa di tempo in trattamento, nel quale i pazienti hanno riportato “fastidio elevato per effetto avverso” (8%) rispetto al braccio di controllo (24%) (IC 95%: -23% -10%, p<0,0001) come valutato dall'item GP5 della Functional Assessment of Cancer Therapy, risposta 3 "Abbastanza" o 4 "Molto".
In una successiva analisi della OS, al data lock dell'11 marzo 2024, sono stati osservati 26 eventi nei due bracci e l'HR è stato di 0,275 (IC 95%: 0,124; 0,608). L'HR della PFS per questa analisi è stato di 0,202 (IC 95%: 0,128, 0,320) e l'ORR per selpercatinib è stato dell'82,4% rispetto al 43,9% per il braccio di controllo.
LIBRETTO-001
Tra i 324 pazienti con MTC con mutazione di RET arruolati nello studio LIBRETTO-001, 143 erano naïve al trattamento con cabozantinib e vandetanib. Di questi, 116 erano naïve ad altre terapie sistemiche e 27 avevano ricevuto in precedenza altre terapie sistemiche. Tra i pazienti naïve a cabozantinib e vandetanib, l’età mediana era 57 anni (intervallo (range) tra 15 e 87 anni). 2 pazienti (1,4%) erano di età < 18 anni. Il 58, 0% dei pazienti erano di sesso maschile. La categorizzazione dei pazienti è stata: 86,7% bianchi, il 5,6% asiatici e l’1,4% neri. Al momento dell’arruolamento, la maggior parte dei pazienti (97,9%) presentava malattia metastatica. L’ECOG performance status è stato riportato come 0-1 (95,9%) o 2 (4,2%). La mutazione più comune era M918T (60,1%), seguita dalle mutazioni di cisteina extracellulare (23,8%). I risultati di efficacia per il trattamento di pazienti con MTC con mutazione di RET naïve al trattamento con cabozantinib e vandetanib, sono riassunti nella tabella 13.
Tabella 13 LIBRETTO-001 Risposte obiettive e durata della risposta
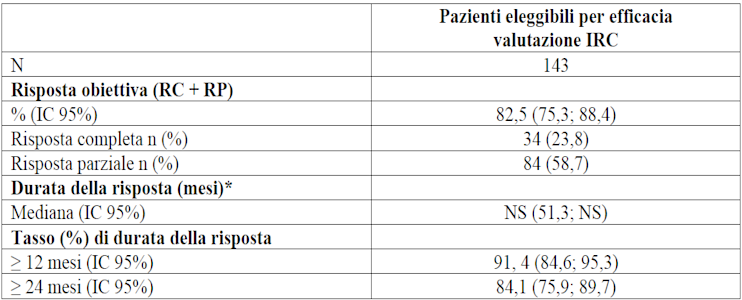
IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile, RP = risposta parziale
*La durata mediana del follow up è stata di 39,4 mesi (25°, 75° percentile: 32,3; 45,4).
Data di cut-off dei dati: 13 gennaio 2023
Cancro midollare della tiroide con mutazione di RET- pretrattato
Tra i pazienti con MTC con mutazione di RET arruolati nello studio LIBRETTO-001, 152 erano stati precedentemente trattati con cabozantinib e/o vandetanib e considerati eleggibili per la valutazione di efficacia. L’età mediana era di 58 anni (intervallo (range) tra 17 e 90 anni); 1 paziente (0,7%) aveva un’età < 18 anni. Il 63,8% dei pazienti era di sesso maschile. La categorizzazione dei pazienti è stata: 90,1% bianchi, mentre l’1,3% asiatici e l’1,3% neri. L’ECOG performance status è stato riportato come 0-1 (92,7%) o 2 (7,2%). Il 98,0% dei pazienti aveva patologia metastatica. La mutazione più comune era M918T (65,1%), seguita da mutazioni della cisteina extracellulare (15,8%), il 100% (n = 152) dei pazienti aveva ricevuto una precedente terapia sistemica con una mediana di 2 precedenti regimi sistemici e il 27,6% (n = 42) aveva ricevuto 3 o più regimi sistemici.
I risultati di efficacia nel MTC con mutazione di RET precedentemente trattato sono riassunti nella tabella 14.
Tabella 14 LIBRETTO-001 Risposta obiettiva e durata della risposta
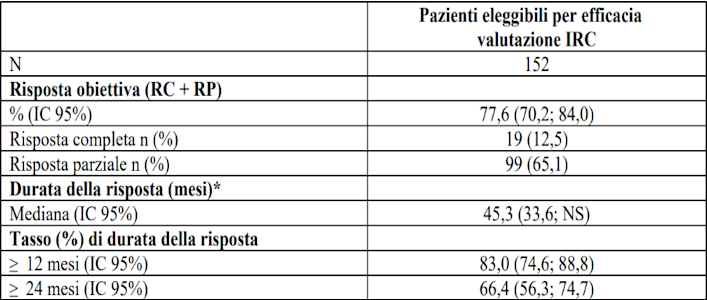
IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile, RP = risposta parziale
*La durata mediana del follow up è stata 38,3 mesi (25°, 75° percentile:23,0; 46,1).
Data di cut-off dei dati: 13 gennaio 2023
Altri tumori solidi RET fusione-positivi
L'efficacia è stata valutata in 75 pazienti con tumori RET fusione-positivi diversi dal NSCLC e dal cancro della tiroide con progressione della malattia durante o dopo un precedente trattamento sistemico o che non avevano opzioni terapeutiche alternative soddisfacenti. L'età mediana era di 59 anni (intervallo (range) da 21 a 92); il 50,7% erano donne; il 60,0% erano bianchi, il 34,7% asiatici e il 4,0% neri; l’ECOG performance status è stato di 0-1 (90,6%) o 2 (9,3%) e il 96,0% dei pazienti presentava una malattia metastatica. Sessantanove pazienti (92,0%) hanno ricevuto una precedente terapia sistemica con una mediana di 2 terapie sistemiche precedenti (intervallo (range) da 0 a 9) e il 36,0% ha ricevuto 3 o più terapie sistemiche precedenti. Nessun paziente era stato precedentemente
trattato con un inibitore selettivo di RET. I tumori più comuni sono stati colon (29,3%), tumore pancreatico (24,0%), salivare (6,7%), sarcoma (6,7%) e colangiocarcinoma (6,7%). I partner di fusione più comuni sono stati NCOA4 (38,7%), CCDC6 (20,0%) e KIF5B (8,0%). I risultati di efficacia per i tumori solidi RET fusione-positivi diversi dal NSCLC e dal cancro della tiroide sono riassunti nella Tabella 15 e nella Tabella 16.
Tabella 15 LIBRETTO-001 Risposta obiettiva e durata della risposta
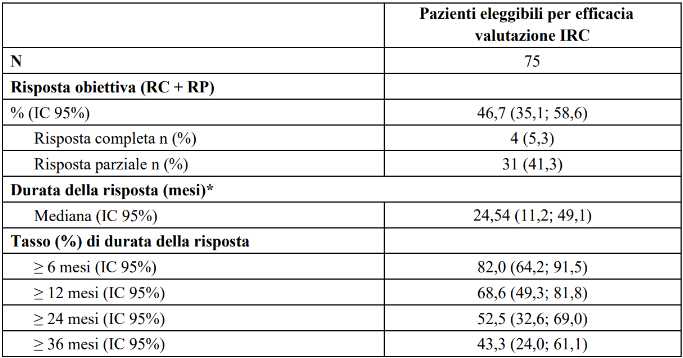
*La durata mediana del follow up è stata 32,23 mesi (25°, 75° percentile:13,3; 50,8).
IC = Intervallo di confidenza, RC = risposta completa, NS = non stimabile, RP = risposta parziale
Data di cut-off dei dati: 14 febbraio 2025
Tabella 16 LIBRETTO-001 Risposta obiettiva e durata della risposta per tipo di tumore
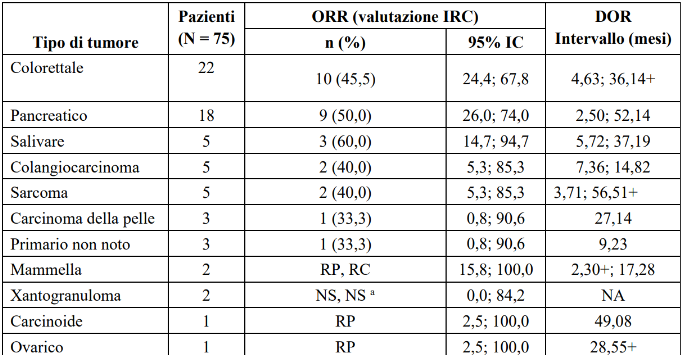
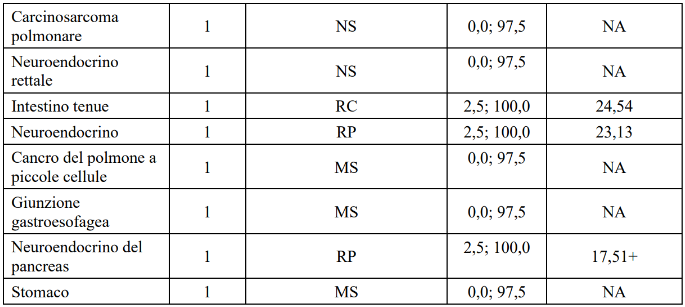
+ indica una risposta in corso.
a Un paziente con xantogranuloma presentava una malattia che non poteva essere valutata dall'IRC a causa del fatto che la pelle era l'unico sito di malattia. Sulla base della valutazione dello sperimentatore, questo paziente ha avuto una RC.
IC = intervallo di confidenza, RC = risposta completa, DOR = durata della risposta, NA = non applicabile, NS = non stimabile, ORR = tasso di risposta obiettiva, RP = risposta parziale, MS= malattia stabile.
Data di cut-off dei dati: 14 febbraio 2025
A causa della rarità del cancro RET fusione-positivo, i pazienti sono stati studiati in più tipi di tumore con un numero limitato di pazienti in alcuni tipi di tumore, causando incertezza nella stima dell'ORR, per tipo di tumore. L'ORR nella popolazione totale può non riflettere la risposta attesa in uno specifico tipo di tumore.
Popolazione pediatrica
Alla data dell’8 novembre 2024, l’efficacia è stata valutata in 36 pazienti di età compresa tra 2 e ≤ 20 anni trattati nello studio LIBRETTO-121, uno studio di fase 1/2 in pazienti pediatrici con un tumore avanzato solido o primario del SNC esprimente un’alterazione attivante di RET. Di questi 36 pazienti, 31 avevano meno di 18 anni di età. Dei 36 pazienti, 14 pazienti presentavano MTC con mutazione RET, 1 paziente presentava MTC positivo per fusione RET, 15 pazienti presentavano carcinoma tiroideo positivo per fusione RET, 3 pazienti presentavano altri tumori solidi positivi per fusione RET e 3 pazienti presentavano altri tumori solidi con mutazione RET.
Dei 14 pazienti con MTC con mutazione RET, 11 pazienti avevano meno di 18 anni, di cui 5 avevano meno di 12 anni di età. Per tutti i 14 pazienti, secondo IRC, la percentuale di risposta obiettiva è stata del 57,1 % (IC 95 %: 28,9; 82,3). 5 pazienti hanno presentato una risposta completa confermata mentre 3 pazienti hanno presentato una risposta parziale confermata.
Per i 14 pazienti, la durata mediana della risposta (mesi) non era stimabile (IC al 95 %: 24,9; NS). La percentuale (%) di pazienti con una durata della risposta ≥ 24 mesi è stata del 100 % (IC al 95 %: 100; 100) e a ≥ 30 mesi è stata dell’83,3 % (IC al 95 %: 27,3; 97,5).
Dei 15 pazienti con carcinoma tiroideo positivo per fusione RET, 13 pazienti avevano meno di 18 anni, di cui 3 avevano meno di 12 anni di età. Dei 15 pazienti, 7 sono stati precedentemente trattati con iodio radioattivo. Per tutti i 15 pazienti, secondo IRC, la percentuale di risposta obiettiva è stata del 53,3 % (IC al 95 %: 26,6; 78,7). 4 pazienti hanno avuto una risposta completa confermata mentre 4 pazienti hanno avuto una risposta parziale confermata. Per i 15 pazienti, la durata mediana della risposta (mesi) non era stimabile (IC al 95 %: NS; NS). La percentuale (%) di pazienti con una durata della risposta ≥ 42 mesi è stata del 100 % (IC al 95 %: 100; 100).
Dei 3 pazienti con altri tumori solidi positivi per fusione RET, tutti e 3 avevano meno di 18 anni, 1 dei quali aveva meno di 12 anni di età. Dei 3 pazienti, 2 presentavano una risposta parziale confermata.
Dei 3 pazienti con altri tumori solidi con mutazione RET, tutti e 3 avevano meno di 18 anni, di questi 2 avevano meno di 12 anni di età. Dei 3 pazienti con altri tumori solidi con mutazione RET, nessun paziente ha avuto una risposta al trattamento con selpercatinib.
L’Agenzia europea per i medicinali ha previsto l’esonero dall’obbligo di presentare i risultati degli studi con selpercatinib nei pazienti di età pari o inferiore a 6 mesi con tumori solidi (vedere paragrafo 4.2 per informazioni sull’uso pediatrico).
5.2 Proprietà farmacocinetiche
Le proprietà farmacocinetiche di selpercatinib sono state valutate in pazienti con tumore solido localmente avanzato o metastatico a cui è stato somministrato selpercatinib due volte al giorno alla dose di 160 mg, salvo diversamente specificato. L’AUC e Cmax di selpercatinib allo stato stazionario sono aumentate in maniera lineare a più che proporzionale alla dose, nell’intervallo di dose compreso tra 20 mg una volta al giorno e 240 mg due volte al giorno.
Lo stato stazionario è stato raggiunto approssimativamente entro 7 giorni e il tasso di accumulo mediano dopo somministrazione di 160 mg di selpercatinib due volte al giorno è stato di 3,4 volte. La Cmax mediana di selpercatinib allo stato stazionario [coefficiente di variazione (CV%)] è stata 2 980 (53%) ng/mL e l’AUC0-24h è stata 51 600 (58%) ng*h/mL.
Gli studi in vivo indicano che selpercatinib è un inibitore lieve della P-gp.
Studi in vitro indicano che selpercatinib non inibisce o induce CYP1A2, CYP2B6, CYP2C9, CYP2C19 o CYP2D6 a concentrazioni clinicamente rilevanti.
Studi in vitro indicano che selpercatinib inibisce MATE1 e BCRP, ma non inibisce OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BSEP e MATE2-K a concentrazioni clinicamente rilevanti. Selpercatinib può aumentare la creatinina sierica diminuendo la secrezione renale tubulare della creatinina attraverso l’inibizione di MATE1.
Le forme farmaceutiche di selpercatinib in capsule rigide e compresse rivestite con film sono bioequivalenti.
Assorbimento
Dopo una dose orale di 160 mg, Retsevmo è stato rapidamente assorbito con tmax approssimativamente di 2 ore. La media geometrica della biodisponibilità assoluta di selpercatinib è stata del 73,2% (intervallo: 60,2-81,5%),
Effetto del cibo
Rispetto all’AUC e Cmax di selpercatinib somministrato a digiuno, l’AUC di selpercatinib è aumentata del 9% e la Cmax ridotta del 14% dopo somministrazione orale a pazienti sani, di una dose singola da 160 mgassunta insieme ad un pasto ricco di grassi. Questi cambiamenti non sono stati considerati clinicamente rilevanti. Pertanto selpercatinib può essere assunto con o senza cibo.
Distribuzione
Il volume di distribuzione (Vss/F) medio apparente (CV%) di selpercatinib stimato dall’analisi di farmacocinetica di popolazione è di 203,1 L (69%) a seguito di somministrazione orale di selpercatinib in pazienti adulti. Selpercatinib in vitro è legato per il 96% alle proteine plasmatiche dell’uomo e il legame è indipendente dalla concentrazione. Il rapporto di concentrazione sangue-plasma è dello 0,7.
Biotrasformazione
Selpercatinib è metabolizzato prevalentemente dal CYP3A4. Dopo somministrazione orale di una dose singola di 160 mg di selpercatinib radiomarcato con [14C] in soggetti sani, selpercatinib immodificato costituiva l’86% dei componenti radioattivi misurati nel plasma.
Eliminazione
La clearance media apparente (CV%) di selpercatinib (CL/F) è di 5,5 L/h (45%) e l’emivita è di 26,5 ore dopo somministrazione orale di selpercatinib in pazienti adulti.
Dopo somministrazione orale di una dose singola di 160 mg di selpercatinib radiomarcato con [14C] in soggetti sani, il 69% (14% immodificata) della radioattività è stato escreto per via fecale e il 24% (11,5% immodificato) è stato trovato nelle urine.
Popolazioni particolari
Età, sesso e peso corporeo
L’età (intervallo tra 2 e 92 anni) o il sesso non hanno avuto un impatto significativo sulla farmacocinetica di Retsevmo. Il Vss/F e la CL/F di selpercatinib aumentano con l’aumentare del peso corporeo (da 9,6 kg a 179 kg).
I pazienti di età pari o superiore a 12 anni con un peso corporeo < 50 kg devono iniziare il trattamento con Retsevmo con una dose di 120 mg due volte al giorno, mentre i pazienti di peso ≥ 50 kg devono iniziare il trattamento con Retsevmo con una dose di 160 mg due volte al giorno.
I pazienti pediatrici di età compresa tra 2 e meno di 12 anni devono iniziare il trattamento con Retsevmo a una dose determinata dalla loro superficie corporea (Tabella 1). Retsevmo non è raccomandato per i pazienti pediatrici con superficie corporea inferiore a 0,45 m2
.
Compromissione epatica
L’AUC0-∞ di selpercatinib è aumentata del 7% in soggetti con classificazione Child-Pugh lieve, del 32% in soggetti con classificazione moderata. Pertanto l’esposizione (AUC) a selpercatinib in soggetticon compromissione epatica lieve o moderata (Child-Pugh classe A e B) è paragonabileall’esposizione in soggetti sani quando viene somministrata una dose di 160 mg.
L’AUC0-∞ di selpercatinib è aumentata del 77% nei soggetti con compromissione epatica severa (Child-Pugh classe C). Esistono dati limitati sulla sicurezza di selpercatinib in pazienti con compromissione epatica severa. Per questo motivo è raccomandata la modifica della dose nei pazienti con compromissione epatica severa (paragrafo 4.2).
Compromissione renale
In uno studio di farmacologia che usava dosi singole di selpercatinib 160 mg, l’esposizione (AUC) era immodificata in soggetti con compromissione renale lieve, moderata o severa. Nefropatia allo stadio terminale (eGFR < 15 mL/min) e pazienti dializzati non sono stati studiati.
Popolazione pediatrica
Le esposizioni di selpercatinib nei pazienti pediatrici di età pari o superiore a 2 anni sono paragonabili a quelle dei pazienti adulti trattati con i dosaggi raccomandati.
5.3 Dati preclinici di sicurezza
Studi a dosi ripetute sono stati condotti su ratti giovani e adolescenti/adulti e su maialini adolescenti/adulti, per caratterizzare la tossicità. Gli organi target della tossicità comuni a ratti e maialini sono stati il sistema ematopoietico, i tessuti linfoidi, la lingua, il pancreas, il tratto gastrointestinale, le cartilagini di crescita epifisarie e i tessuti riproduttivi maschili. In generale, le tossicità in questi organi erano reversibili, eccezioni sono state la tossicità testicolare negli animali adolescenti/adulti e giovani e i cambiamenti nelle cartilagini di accrescimento nei ratti giovani. È stata osservata tossicità reversibile a carico delle ovaie dei soli maialini. Ad alte dosi, la tossicità gastrointestinale nei maialini ha causato morbilità alle esposizioni che erano in genere più basse rispetto alle esposizioni determinate negli uomini alla dose raccomandata. In uno studio sul maialino, le femmine hanno mostrato un incremento lieve e reversibile nel prolungamento del QTc approssimativamente del 12% rispetto ai controlli e del 7% rispetto ai valori pre-dose. Gli organi target della tossicità osservati solo nei ratti sono stati i denti incisivi, il fegato, la vagina, i polmoni, la ghiandola di Brunner e la mineralizzazione multi-tessuto associata con iperfosfatemia. Queste tossicità, verificatesi esclusivamente in questi organi dei ratti, sono state reversibili.
Tossicità giovanile
L’esposizione a selpercatinib di circa 0,5-2 volte superiore all’esposizione negli uomini adulti ha causato mortalità nei ratti di età inferiore ai 21 giorni. Un’esposizione equiparabile è stata tollerata nei ratti di età pari o superiore a 21 giorni.
Ratti giovani e adolescenti/adulti e maialini adolescenti/adulti con cartilagini di accrescimento aperteai quali è stato somministrato selpercatinib hanno mostrato cambiamenti microscopici di ipertrofia, iperplasia e displasia della cartilagine di accrescimento (fisi). Nei ratti giovani, la displasia delle cartilagini di accrescimento è stata irreversibile e associata a diminuzione della lunghezza del femore e a riduzione della densità minerale ossea. Sono stati osservati cambiamenti scheletrici a livelli di esposizione equivalenti a quelli visti nei pazienti adulti che assumevano la dose raccomandata di 160 mg due volte al giorno.
Ratti maschi giovani ai quali è stato somministrato selpercatinib e ai quali è stato permesso di raggiungere l’età riproduttiva dopo la cessazione della somministrazione, hanno mostrato diminuzione della performance riproduttiva quando si sono accoppiati con ratti femmine non trattate. Sono stati osservati diminuzione degli indici di fertilità e di accoppiamento, aumento delle perdite pre e post impianto e diminuzione del numero di embrioni vitali ad un'esposizione di circa 3,4 volte l'esposizione efficace negli adulti.
Genotossicità
Selpercatinib non è genotossico alle dosi terapeutiche. In un esperimento in vivo sui saggi del micronucleo nei ratti, selpercatinib era positivo a concentrazioni > 7 volte la Cmax alla dose per l’uomo di 160 mg due volte al giorno. In un esperimento in vitro sui saggi del micronucleo nei linfociti del sangue periferico umano, è stata osservata una risposta ambigua alla concentrazione di circa 485 volte la Cmax della dose per l’uomo.
Mutagenesi
Selpercatinib non ha causato mutazioni in un test di mutagenicità batterica.
Carcinogenesi
In uno studio a 2 anni sulla carcinogenicità di selpercatinib nei ratti, sono stati osservati tumori vaginali in alcune femmine a livelli di esposizione plasmatica simili ai livelli osservati nei pazienti adulti trattati con la dose di 160 mg due volte al giorno. Non sono state osservate alterazioni pre-neoplastiche nel tratto riproduttivo di femmine di ratto. La rilevanza clinica di questi risultati non è nota. Selpercatinib non è risultato cancerogeno nei ratti maschi in questo studio.
Selpercatinib non è risultato cancerogeno nei topi maschi e femmine in uno studio a 6 mesi.
Embriotossicità/teratogenicità
Sulla base di dati provenienti da studi sulla riproduzione animale e del meccanismo d’azione, selpercatinib può causare danni fetali quando somministrato a donne in gravidanza. La somministrazione di selpercatinib in ratte gravide durante l’organogenesi con esposizioni materne che erano approssimativamente uguali a quelle osservate alla dose umana raccomandata di 160 mg due volte al giorno hanno dato risultati di embrioletalità e malformazioni.
Tossicità riproduttiva
Risultati di studi condotti sui ratti e maialini suggeriscono che selpercatinib potrebbe compromettere la fertilità negli uomini e nelle donne.
In uno studio di fertilità sui ratti maschi, a livelli di esposizione subclinici basati sull’AUC (0,2 volte l’esposizione clinica alla dose raccomandata per l’uomo), sono stati osservati impoverimento delle cellule germinali e ritenzione degli spermatidi dose-dipendente. Questi effetti sono stati associati a riduzione del peso degli organi, riduzione della motilità spermatica e aumento del numero di spermatozoi anormali a livelli di esposizione basata sull’AUC di circa due volte l’esposizione clinica alla dose raccomandata per l’uomo. Osservazioni al microscopio nello studio di fertilità sui ratti maschi erano coerenti con gli effetti negli studi a dosi ripetute in ratti e maialini, nei quali degenerazione testicolare dose-dipendente non reversibile è stata associata a riduzione degli spermatozoi luminali nell’epididimo, a seguito di esposizione a livelli subclinici basati sull’AUC (da 0,1 a 0,4 volte l’esposizione clinica alla dose raccomandata per l’uomo).
In uno studio su ratti femmine su fertilità e stadio embrionale precoce, sono stati osservati una riduzione del numero dei cicli estruali così come embrio-letalità a livelli di esposizione basati sull’AUC approssimativamente uguali all’esposizione clinica alla dose raccomandata per l’uomo. Negli studi a dose ripetute nei ratti sono stati osservati mucificazione vaginale reversibile con corneificazione delle singole cellule e cicli estruali alterati a livelli clinicamente rilevanti di esposizione basati sull’AUC. Nei maialini sono stati osservati diminuzione del corpo luteo e/o cisti del corpo luteo a livelli di esposizione subclinica basata su AUC (da 0,07 a 0,3 volte l'esposizione clinica alla dose raccomandata per l’uomo).
6.1 Elenco degli eccipienti
Contenuto della capsula
Cellulosa microcristallina
Silice colloidale anidra
Involucro della capsula
Retsevmo capsule rigide 40 mg
Gelatina
Titanio diossido (E171)
Ossido di ferro (E172)
Retsevmo capsule rigide 80 mg
Gelatina
Titanio diossido (E171)
Blu brillante FCF (E133)
Composizione dell’inchiostro nero delle capsule
Gommalacca
Etanolo (al 96 percento)
Alcol isopropilico
Butanolo
Glicole propilenico
Acqua purificata
Soluzione di ammoniaca, concentrata
Potassio idrossido
Ossido di ferro nero
6.2 Incompatibilità
Non pertinente.
6.3 Periodo di validità
3 anni.
6.4 Precauzioni particolari per la conservazione
Questo medicinale non richiede condizioni di conservazione particolari.
6.5 Natura e contenuto del contenitore
Flacone di plastica
Ciascuna confezione contiene un flacone di polietilene ad alta densità (high-density polyethylene, HDPE) con tappo a vite in plastica.
Retsevmo 40 mg capsule rigide
Retsevmo 40 mg capsule rigide è fornito in flacone HDPE da 60 capsule.
Retsevmo 80 mg capsule rigide
Retsevmo 80 mg capsule rigide è fornito in flacone HDPE da 60 capsule o in flacone HDPE da
120 capsule.
Confezione blister
Retsevmo 40 mg capsule rigide
Fornito come blister di policlorotrifluoroetilene/polivinilcloruro (PCTFE/PVC) sigillati con un foglio di alluminio in un cartoncino per blister, in confezioni da 14, 42, 56 o 168 capsule rigide.
Retsevmo 80 mg capsule rigide
Fornito come blister di PCTFE/PVC sigillati con un foglio di alluminio in un cartoncino per blister, in confezioni da 14, 28, 56 o 112 capsule rigide.
È possibile che non tutte le confezioni siano commercializzate.
6.6 Precauzioni particolari per lo smaltimento
Il medicinale non utilizzato e i rifiuti derivati da tale medicinale devono essere smaltiti in conformità alla normativa locale vigente.
Eli Lilly Nederland B.V.
Papendorpseweg 83
3528BJ Utrecht
Paesi Bassi
EU/1/20/1527/001
EU/1/20/1527/002
EU/1/20/1527/003
EU/1/20/1527/004
EU/1/20/1527/005
EU/1/20/1527/006
EU/1/20/1527/007
EU/1/20/1527/008
EU/1/20/1527/009
EU/1/20/1527/010
EU/1/20/1527/011
Data della prima autorizzazione: 11 febbraio 2021
Data del rinnovo più recente: 13 gennaio 2025
04 giugno 2026
Informazioni più dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia europea per i medicinali https://www.ema.europa.eu.