Per segnalare un effetto indesiderato o un reclamo su un prodotto Lilly, si prega di non utilizzare questo sito web. La segnalazione delle reazioni avverse è estremamente importante. Gli operatori sanitari e i pazienti possono riportare una reazione avversa o un reclamo di un prodotto sul sito di AIFA.
Verzenios® (abemaciclib)
Verzenios 50 mg compresse rivestite con film.
Verzenios 100 mg compresse rivestite con film.
Verzenios 150 mg compresse rivestite con film.
Verzenios 50 mg compresse rivestite con film
Ogni compressa rivestita con film contiene 50 mg di abemaciclib.
Eccipienti con effetti noti
Ogni compressa rivestita con film contiene 14 mg di lattosio monoidrato.
Verzenios 100 mg compresse rivestite con film
Ogni compressa rivestita con film contiene 100 mg di abemaciclib.
Eccipienti con effetti noti
Ogni compressa rivestita con film contiene 28 mg di lattosio monoidrato.
Verzenios 150 mg compresse rivestite con film
Ogni compressa rivestita con film contiene 150 mg di abemaciclib.
Eccipienti con effetti noti
Ogni compressa rivestita con film contiene 42 mg di lattosio monoidrato.
Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
Compresse rivestite con film (compresse).
Verzenios 50 mg compresse rivestite con film
Compressa ovale beige di 5,2 x 9,5 mm, con impresso "Lilly" su un lato e "50" sull'altro.
Verzenios 100 mg compresse rivestite con film
Compressa ovale bianca di 6,6 x 12,0 mm, con impresso "Lilly" su un lato e "100" sull'altro.
Verzenios 150 mg compresse rivestite con film
Compressa ovale gialla di 7,5 x 13,7 mm, con impresso "Lilly" su un lato e "150" sull'altro.
4.1 Indicazioni terapeutiche
Carcinoma mammario in fase iniziale
Verzenios in associazione alla terapia endocrina è indicato per il trattamento adiuvante di pazienti adulti con carcinoma mammario in fase iniziale, positivo al recettore ormonale (HR), negativo al recettore del fattore di crescita umano epidermico di tipo 2 (HER2), linfonodo-positivo, ad alto rischio di recidiva (vedere paragrafo 5.1).
Nelle donne in pre- o perimenopausa, la terapia endocrina con inibitore dell'aromatasi deve essere associata a un agonista dell'ormone di rilascio dell'ormone luteinizzante (LHRH).
Carcinoma mammario avanzato o metastatico
Verzenios è indicato per il trattamento di donne con carcinoma della mammella localmente avanzato o metastatico, positivo ai recettori ormonali (HR), negativo al recettore del fattore umano di crescita epidermico di tipo 2 (HER2) in associazione con un inibitore dell’aromatasi o fulvestrant, come terapia endocrina iniziale, o in donne che hanno ricevuto una precedente terapia endocrina.
Nelle donne in pre- o perimenopausa, la terapia endocrina deve essere associata con un agonista del LHRH.
4.2 Posologia e modo di somministrazione
La terapia con Verzenios deve essere iniziata e condotta con la supervisione di medici esperti nell’uso di terapie antitumorali.
Posologia
La dose raccomandata di abemaciclib è 150 mg due volte al giorno quando utilizzato in associazione con la terapia endocrina. Fare riferimento al riassunto delle caratteristiche del prodotto del medicinale utilizzato come terapia endocrina in associazione per la posologia raccomandata.
Durata del trattamento
Carcinoma mammario in fase iniziale
Verzenios deve essere assunto continuativamente per due anni o fino a quando si verifica una recidiva della malattia o una tossicità inaccettabile.
Carcinoma mammario avanzato o metastatico
Verzenios deve essere assunto in maniera continuativa fino a che la paziente trae beneficio clinico dalla terapia o fino a quando si verifica una tossicità inaccettabile.
Se una paziente ha vomito o salta una dose di Verzenios, la paziente deve essere istruita ad assumere la dose successiva all'orario previsto; non deve essere assunta una dose aggiuntiva.
Modifiche della dose
La gestione di alcuni eventi avversi può richiedere l’interruzione della terapia e/o la riduzione della dose come indicato nelle Tabelle 1-7
Tabella 1. Raccomandazioni per le modifiche della posologia in caso di reazioni avverse.

Tabella 2. Raccomandazioni per la gestione delle tossicità ematologiche.
I parametri ematochimici devono essere monitorati prima dell'inizio della terapia con Verzenios, ogni due settimane per i primi due mesi, mensilmente per i successivi due mesi e come clinicamente indicato. Prima dell’inizio del trattamento, sono raccomandate conte assolute dei neutrofili (ANC) ≥ 1 500/mm3, delle piastrine ≥ 100 000 / mm3 ed emoglobina ≥ 8 g/dL.

a Criteri per una Terminologia Comune degli Eventi Avversi del National Cancer Institute (NCI Common Terminology Criteria for Adverse Events, CTCAE)
b ANC: Grado 1: ANC < LLN - 1 500 / mm3; Grado 2: ANC 1 000 - <1 500 / mm3; Grado 3: ANC 500 - <1000 / mm3; Grado 4: ANC <500 / mm3
LLN = Lower Limit of Normal (limite inferiore di normalità)
Tabella 3. Raccomandazioni per la gestione della diarrea
Il trattamento con farmaci antidiarroici, come loperamide, deve essere iniziato al primo segno di feci molli.

a NCI CTCAE
Tabella 4. Raccomandazioni per la gestione dell’aumento delle transaminasi
L’alanina aminotransferasi (ALT) e l’aspartato aminotransferasi (AST) devono essere monitorate prima dell'inizio della terapia con Verzenios, ogni due settimane per i primi due mesi, mensilmente per i successivi due mesi e come clinicamente indicato.

a NCI CTCAE
ULN = Upper Limit of Normal (limite superiore di normalità)
Tabella 5. Raccomandazioni per la gestione della malattia polmonare interstiziale (ILD –
Interstitial Lung Disease)/polmonite.

a NCI CTCAE
Tabella 6. Raccomandazioni per la gestione di eventi di tromboembolismo venoso (TEV).

a NCI CTCAE
Tabella 7. Raccomandazioni per la gestione delle tossicità non ematologiche (escluse diarrea e aumento delle transaminasi, ILD/polmonite e TEV).

a NCI CTCAE
Inibitori CYP3A4
L'uso concomitante di inibitori potenti del CYP3A4 deve essere evitato. Se gli inibitori potenti del CYP3A4 non possono essere evitati, la dose di abemaciclib deve essere ridotta a 100 mg due volte al giorno.
Nelle pazienti che hanno avuto riduzione della dose a 100 mg di abemaciclib due volte al giorno e nelle quali la co-somministrazione di un inibitore potente del CYP3A4 non può essere evitata, la dose abemaciclib deve essere ulteriormente ridotta a 50 mg due volte al giorno.
Nelle pazienti che hanno avuto riduzione della dose a 50 mg di abemaciclib due volte al giorno e nelle quali non è possibile evitare la co-somministrazione di un inibitore potente del CYP3A4, la dose di abemaciclib può essere proseguita con un attento monitoraggio dei segni di tossicità. In alternativa, la dose di abemaciclib può essere ridotta a 50 mg una volta al giorno o interrotta.
Se l'inibitore del CYP3A4 viene interrotto, la dose di abemaciclib deve essere aumentata fino alla dose utilizzata prima dell'inizio dell'inibitore CYP3A4 (dopo un tempo pari a 3 - 5 emivite dell'inibitore del CYP3A4).
Popolazioni speciali
Pazienti anziani
Non sono richieste modifiche della dose in base all’età (vedere paragrafo 5.2).
Compromissione renale
Non sono necessarie modifiche della dose in pazienti con compromissione renale di grado lieve o moderato. Non ci sono dati riguardanti la somministrazione di abemaciclib in pazienti con compromissione renale severa, malattia renale allo stadio terminale o in pazienti dializzate (vedere paragrafo 5.2). Abemaciclib deve essere usato con cautela in pazienti con compromissione renale severa, con un attento monitoraggio dei segni di tossicità.
Compromissione epatica
Non sono necessarie modifiche della dose in pazienti con compromissione epatica di grado lieve (Child Pugh A) o moderato (Child Pugh B). In pazienti con compromissione epatica severa (Child Pugh C), è raccomandata la diminuzione della frequenza della dose a una volta al giorno (vedere paragrafo 5.2).
Popolazione pediatrica
La sicurezza e l’efficacia di abemaciclib nei bambini e negli adolescenti di età inferiore a 18 anni non sono state stabilite. Non ci sono dati disponibili.
Modo di somministrazione
Verzenios è per uso orale.
La dose può essere assunta con o senza cibo. Non deve essere assunto con pompelmo o succo di pompelmo (vedere paragrafo 4.5).
Le pazienti devono assumere le dosi all'incirca alla stessa ora ogni giorno.
La compressa deve essere deglutita intera (le pazienti non devono masticare, rompere o dividere le compresse prima di deglutirle).
4.3 Controindicazioni
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
4.4 Avvertenze speciali e precauzioni d’impiego
Neutropenia
La neutropenia è stata riportata in pazienti che hanno ricevuto abemaciclib. È raccomandata la modifica della dose per le pazienti che sviluppano neutropenia di Grado 3 o 4 (vedere paragrafo 4.2). Eventi fatali di sepsi neutropenica si sono verificati in < 1 % delle pazienti con carcinoma mammario metastatico. Le pazienti devono essere istruite a segnalare al loro medico qualsiasi episodio di febbre.
Infezioni/infestazioni
Nelle pazienti in trattamento con abemaciclib più terapia endocrina è stata riportata una percentuale più alta di infezioni rispetto alle pazienti trattate con terapia endocrina. L’infezione polmonare senza neutropenia concomitante è stata riportata in pazienti che hanno ricevuto abemaciclib. Eventi fatali si sono verificati in < 1 % delle pazienti con carcinoma mammario metastatico.Le pazienti devono essere monitorate per segni e sintomi di infezioni e trattate come clinicamente appropriato.
Tromboembolismo venoso
Eventi tromboembolici venosi sono stati riportati nelle pazienti trattate con abemacicib più terapia endocrina. Le pazienti devono essere monitorate per segni e sintomi di trombosi venosa profonda ed embolia polmonare e trattate come clinicamente appropriato. In base al grado di TEV, abemaciclib può richiedere la modifica della dose (vedere paragrafo 4.2).
Eventi tromboembolici arteriosi
Negli studi nel carcinoma mammario metastatico è stato osservato un potenziale aumento del rischio di eventi tromboembolici arteriosi gravi (ETA), inclusi ictus ischemico e infarto miocardico, quando abemaciclib è stato somministrato in associazione con terapie endocrine. Devono essere presi in considerazione i benefici e i rischi della continuazione di abemaciclib nelle pazienti che manifestano un ETA grave.
Aumento delle transaminasi
Sono stati riportati aumenti di ALT e AST in pazienti trattate con abemaciclib. In base all’aumento dei livelli di ALT o AST, abemaciclib può richiedere una modifica della dose (vedere paragrafo 4.2).
Diarrea
La diarrea è la reazione avversa più comune. In tutti gli studi clinici, il tempo mediano di comparsa del primo evento di diarrea è stato di circa 6-8 giorni, e la durata mediana della diarrea è stata da 7 a 12 giorni (Grado 2) e da 5 a 8 giorni (Grado 3). La diarrea può essere associata a disidratazione. Le pazienti devono iniziare il trattamento con agenti antidiarroici come loperamide al primo segno di feci molli, aumentare l’assunzione di liquidi per via orale e informare il proprio medico. La modifica della dose è raccomandata per pazienti che sviluppano diarrea ≥ Grado 2 (vedere paragrafo 4.2)
ILD/polmonite
ILD/polmonite sono state riportate in pazienti in trattamento con abemaciclib. I pazienti devono essere monitorati per i sintomi polmonari indicativi di ILD/polmonite e trattati come clinicamente appropriato. In base al grado di ILD/polmonite, può essere necessario modificare il dosaggio di abemaciclib (vedere paragrafo 4.2). Interrompere permanentemente abemaciclib in pazienti con ILD/polmonite di Grado 3 o 4.
Uso concomitante di induttori del CYP3A4
L'uso concomitante di induttori del CYP3A4 deve essere evitato a causa del rischio di ridotta efficacia di abemaciclib (vedere paragrafo 4.5).
Crisi viscerale
Non ci sono dati sull'efficacia e la sicurezza di abemaciclib in pazienti con crisi viscerale.
Lattosio
Le pazienti con rari problemi ereditari di intolleranza al galattosio, deficit totale di lattasi o
malassorbimento di glucosio-galattosio non devono assumere questo medicinale.
Sodio
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per compressa, cioè essenzialmente “senza sodio”.
4.5 Interazioni con altri medicinali ed altre forme d’interazione
Effetti di altri medicinali sulla farmacocinetica di abemaciclib
Abemaciclib è metabolizzato principalmente dal CYP3A4.
Inibitori del CYP3A4
La co-somministrazione di abemaciclib con gli inibitori del CYP3A4 può aumentare le concentrazioni plasmatiche di abemaciclib. Nelle pazienti con carcinoma in fase avanzata e/o metastatico, la co-somministrazione dell’inibitore del CYP3A4 claritromicina ha determinato un aumento dell’esposizione plasmatica ad abemaciclib di 3,4 volte e un aumento di 2,5 volte dell'esposizione plasmatica combinata di abemaciclib e dei suoi metaboliti attivi, corretta per la potenza riferita alla frazione libera.
L’uso di inibitori potenti del CYP3A4 insieme ad abemaciclib deve essere evitato. Se è necessaria la co-somministrazione di inibitori potenti del CYP3A4, la dose di abemaciclib deve essere ridotta (vedere paragrafo 4.2), seguita da un attento monitoraggio della tossicità. Esempi di inibitori potenti del CYP3A4 includono, ma non si limitano a: claritromicina, itraconazolo, ketoconazolo, lopinavir/ritonavir, posaconazolo o voriconazolo. Evitare il pompelmo o il succo di pompelmo.
Non è necessario alcun aggiustamento della dose per le pazienti trattate con inibitori del CYP3A4 deboli o moderati. Tuttavia, devono essere attentamente monitorati i segni di tossicità.
Induttori del CYP3A4
La co-somministrazione di abemaciclib con l’induttore potente del CYP3A4 rifampicina ha
determinato una riduzione della concentrazione plasmatica di abemaciclib del 95 % e una riduzione del 77 % dell'AUC0-∞ di abemaciclib e dei suoi metaboliti attivi, corretta per la potenza riferita alla frazione libera. L'uso concomitante di induttori potenti del CYP3A4 (inclusi, ma non limitati a: carbamazepina, fenitoina, rifampicina e erba di San Giovanni) deve essere evitato a causa del rischio di ridotta efficacia di abemaciclib.
Effetti di abemaciclib sulla farmacocinetica di altri medicinali
Medicinali che sono substrato di trasportatori
Abemaciclib e i suoi principali metaboliti attivi inibiscono i trasportatori renali, come il trasportatore dei cationi organici 2 (OCT2), la proteina di estrusione multifarmaco e di tossine (MATE1) e la MATE2-K. In vivo, possono verificarsi interazioni di abemaciclib con substrati clinicamente rilevanti di questi trasportatori, come dofetilide o creatinina (vedere paragrafo 4.8). In uno studio clinico di interazione farmacologica con metformina (substrato di OCT2, MATE1 e 2) co-somministrata con 400 mg di abemaciclib, è stato osservato un piccolo aumento (37 %) ma non clinicamente rilevante dell'esposizione plasmatica della metformina. Questo è risultato essere dovuto ad una ridotta secrezione renale con filtrazione glomerulare inalterata.
Nei soggetti sani, la co-somministrazione di abemaciclib e di loperamide, substrato della glicoproteina P (P-gp), ha determinato un aumento dell'esposizione plasmatica di loperamide del 9 % dell’AUC0-∞ e el 35 % della Cmax. Questo non è stato considerato clinicamente rilevante. Tuttavia, sulla base dell'inibizione in vitro della P-gp e della proteina di resistenza del tumore mammario (BCRP) osservate con abemaciclib, in vivo possono verificarsi interazioni di abemaciclib con i substrati di questi trasportatori aventi un indice terapeutico ristretto, come digossina o dabigatran etexilato.
In uno studio clinico condotto su pazienti con carcinoma mammario non è stata osservata
un'interazione farmacocinetica clinicamente rilevante tra abemaciclib e anastrozolo, fulvestrant, exemestane, letrozolo o tamoxifene.
Attualmente non è noto se abemaciclib possa ridurre l'efficacia dei contraccettivi ormonali ad azione sistemica.
4.6 Fertilità, gravidanza e allattamento
Donne in età fertile/contraccezione nelle donne
Le donne in età fertile devono usare un metodo contraccettivo altamente efficace (ad es. una
contraccezione a doppia barriera) durante il trattamento e per almeno 3 settimane dopo il
completamento della terapia (vedere paragrafo 4.5).
Gravidanza
Non ci sono dati sull'uso di abemaciclib in donne in gravidanza. Gli studi su animali hanno mostrato tossicità riproduttiva (vedere paragrafo 5.3). Verzenios non è raccomandato durante la gravidanza e nelle donne in età fertile che non facciano uso di misure contraccettive.
Allattamento
Non è noto se abemaciclib sia escreto nel latte umano. Non è possibile escludere un rischio per i bambini allattati al seno. Le pazienti che assumono abemaciclib non devono allattare.
Fertilità
L'effetto di abemaciclib sulla fertilità negli esseri umani non è noto. Mentre nei ratti non sono stati osservati effetti sulla fertilità maschile, gli effetti citotossici sul tratto riproduttivo maschile nei topi, ratti e cani indicano che abemaciclib può compromettere la fertilità nei maschi. Non sono stati osservati effetti avversi sugli organi riproduttivi femminili nei topi, ratti o cani, né effetti sulla fertilità femminile e sullo sviluppo embrionale precoce nei ratti (vedere paragrafo 5.3)
4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Verzenios altera lievemente la capacità di guidare veicoli e di usare macchinari. Le pazienti devono essere informate di prestare attenzione durante la guida o l'uso di macchinari nel caso in cui manifestino stanchezza o capogiro durante il trattamento con Verzenios (vedere paragrafo 4.8).
4.8 Effetti indesiderati
Riassunto del profilo di sicurezza
Le reazioni avverse più comuni sono diarrea, infezioni, neutropenia, leucopenia, anemia, stanchezza, nausea, vomito, alopecia e appetito ridotto.
Tra le reazioni avverse più comuni, gli eventi di Grado ≥ 3 sono stati inferiori al 5 % ad eccezione di neutropenia, leucopenia e diarrea.
Tabella delle reazioni avverse
Nelle seguenti tabelle, le reazioni avverse sono elencate in base alla classificazione per sistemi e organi e frequenza secondo MedDRA. Le classificazioni di frequenza sono: molto comune ≥ 1 / 10, comune (da ≥ 1 / 100 a < 1 / 10), non comune (da ≥ 1 / 1 000 a < 1 / 100), raro (da ≥ 1 / 10 000 a < 1 / 1 000), molto raro (< 1 / 10 000) e non noto (la frequenza non può essere stimata sulla base dei dati disponibili).
All’interno di ogni gruppo di frequenza, le reazioni avverse sono elencate in ordine di gravità decrescente.
Tabella 8. Reazioni averse riportate negli studi di fase 3 di abemaciclib in associazione con la terapia endocrinaa (N = 3 559) e dopo l’immissione in commercio
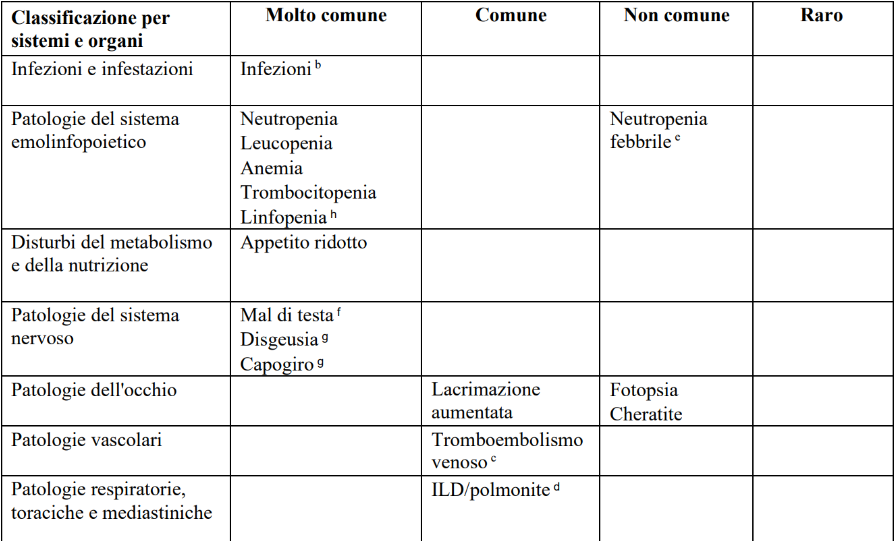
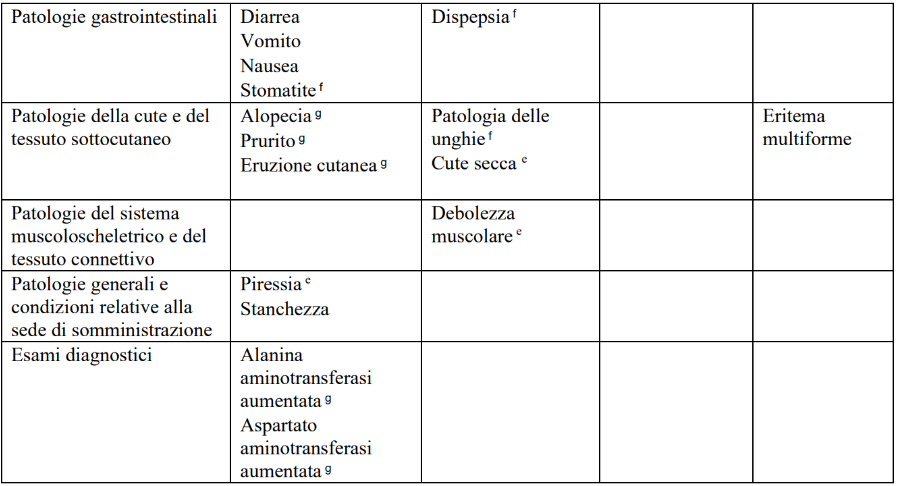
a Abemaciclib in associazione con anastrozolo, letrozolo, exemestane, tamoxifene, o fulvestrant.
b Infezioni includono tutti i termini preferiti riportati che sono parte del gruppo infezioni e infestazioni della classificazione per sistemi e organi.
c Eventi tromboembolici venosi includono trombosi venosa profonda (TVP), embolismo polmonare, trombosi del seno venoso cerebrale, trombosi della vena ascellare e della succlavia, TVP della vena cava inferiore e trombosi venosa pelvica.
d ILD/polmonite per il carcinoma mammario in fase iniziale (early breast cancer -EBC) includono tutti i termini preferiti riportati che sono parte dell'SMQ MedDRA malattia polmonare interstiziale. Per il carcinoma mammario metastatico (metastatic breast cancer - mBC) i termini preferiti includono malattia polmonare interstiziale, polmonite, polmonite organizzativa, fibrosi polmonare e bronchiolite obliterante.
e ADR (Adverse Drug Reaction, reazioni avverse) considerate solo nel contesto mBC (MONARCH 2 e MONARCH 3).
f ADR considerate solo nel contesto EBC (monarchE).
g Frequenza comune nel contesto EBC (monarchE), molto comune nel contesto mBC (MONARCH 2 e MONARCH 3).
h Frequenza comune nel contesto mBC (MONARCH 2 e MONARCH 3), molto comune nel contesto EBC (monarchE).
Descrizione di reazioni avverse selezionate
Neutropenia
La neutropenia è stata segnalata frequentemente in tutti gli studi. Nello studio monarchE, la
neutropenia è stata segnalata nel 45,8 % dei pazienti. Una diminuzione di Grado 3 o 4 della conta dei neutrofili (sulla base dei risultati di laboratorio) è stata segnalata nel 19,1 % dei pazienti trattati con abemaciclib in associazione a terapia endocrina con un tempo mediano di insorgenza di 30 giorni e un tempo mediano di risoluzione di 16 giorni. La neutropenia febbrile è stata segnalata nello 0,3 % dei pazienti. Negli studi MONARCH 2 e MONARCH 3, la neutropenia è stata segnalata nel 45,1 % dei pazienti. Una diminuzione di Grado 3 o 4 della conta dei neutrofili (sulla base dei risultati di laboratorio) è stata segnalata nel 28,2 % delle pazienti trattate con abemaciclib in associazione con inibitori dell'aromatasi o fulvestrant. Il tempo mediano di insorgenza della neutropenia di Grado 3 o 4 era da 29 a 33 giorni e il tempo mediano per la risoluzione era di 11-15 giorni. La neutropenia febbrile è stata riportata nello 0,9 % dei pazienti. È raccomandata la modifica della dose per le pazienti che sviluppano neutropenia di Grado 3 o 4 (vedere paragrafo 4.2).
Diarrea
La diarrea è stata la reazione avversa riportata più comunemente (vedere Tabella 8). L’incidenza è stata maggiore durante il primo mese di trattamento con abemaciclib e si è ridotta successivamente. Nello studio monarchE, il tempo mediano di insorgenza del primo evento di diarrea di qualsiasi grado è stato di 8 giorni. La durata mediana della diarrea è stata di 7 giorni per il Grado 2 e di 5 giorni per il Grado 3. Negli studi MONARCH 2 e MONARCH 3, il tempo mediano di insorgenza del primo evento di diarrea di qualsiasi grado è stato approssimativamente da 6 a 8 giorni. La durata mediana della diarrea è stata da 9 a 12 giorni per il Grado 2 e da 6 a 8 giorni per il Grado 3. La diarrea si è risolta o è passata ad un grado inferiore con un trattamento di supporto come loperamide e/o aggiustamento della
dose (vedere paragrafo 4.2).
Aumento delle transaminasi
Nello studio monarchE, sono stati riportati frequentemente aumenti di ALT e AST (rispettivamente 12,3 % e 11,8 %) in pazienti che ricevevano abemaciclib in associazione con terapia endocrina. Aumenti di ALT o AST di Grado 3 o 4 (sulla base dei risultati di laboratorio) sono stati riportati nel 2,6 % e nell'1,6 % dei pazienti. Il tempo mediano di insorgenza dell'aumento delle ALT di Grado 3 o 4 è stato di 118 giorni e il tempo mediano di risoluzione è stato di 14,5 giorni. Il tempo mediano di insorgenza dell'aumento delle AST di Grado 3 o 4 è stato di 90,5 giorni e il tempo mediano di risoluzione è stato di 11 giorni. Negli studi MONARCH 2 e MONARCH 3, sono stati riportati frequentemente aumenti di ALT e AST (rispettivamente 15,1 % e 14,2 % ) nelle pazienti trattate con abemaciclib in associazione a inibitori dell'aromatasi o fulvestrant. Aumenti delle ALT o AST di Grado 3 o 4 (sulla base dei risultati di laboratorio) sono stati riportati nel 6,1 % e nel 4,2 % delle pazienti. Il tempo mediano di insorgenza dell'aumento di ALT di Grado 3 o 4 è stato da 57 a 61 giorni
e il tempo mediano di risoluzione è stato di 14 giorni. Il tempo mediano di insorgenza dell'aumento di AST di Grado 3 o 4 è stato da 71 a 185 giorni e il tempo mediano di risoluzione è stato da 13 a 15 giorni. È raccomandata la modifica della dose per le pazienti che sviluppano un aumento di ALT o AST di Grado 3 o 4 (vedere paragrafo 4.2).
Creatinina
Sebbene non sia una reazione avversa, è stato dimostrato che abemaciclib aumenta la creatinina sierica. Nello studio monarchE, il 99,3 % dei pazienti ha presentato aumenti della creatinina sierica (sulla base dei risultati di laboratorio) e di questi, lo 0,5 % dei pazienti presentava aumenti di Grado 3 o 4. Nei pazienti che ricevevano la sola terapia endocrina, il 91,0 % ha riportato un aumento della creatinina sierica (di qualsiasi grado di laboratorio). Negli studi MONARCH 2 e MONARCH 3, il 98,3 % delle pazienti presentava aumenti della creatinina sierica (sulla base dei risultati di laboratorio), e di questi l’1,9 % dei pazienti ha presentato aumenti di Grado 3 o 4. Nelle pazienti trattate con un inibitore dell'aromatasi o fulvestrant da soli, il 78,4 % ha riportato un aumento della creatinina sierica (di qualsiasi grado di laboratorio). È stato dimostrato che abemaciclib aumenta la creatinina sierica a
causa dell'inibizione dei trasportatori della secrezione tubulare renale senza alterare la funzione glomerulare (misurata mediante clearance di iohexolo) (vedere paragrafo 4.5). Negli studi clinici, l'aumento della creatinina sierica si è verificato entro il primo mese di somministrazione di abemaciclib, è rimasto elevato ma stabile durante il periodo di trattamento, è stato reversibile dopo l’interruzione del trattamento e non è stato accompagnato da cambiamenti nei marcatori della funzionalità renale, come l'azoto ureico ematico (BUN – blood urea nitrogen), la cistatina C o la velocità di filtrazione glomerulare calcolata sulla base della cistatina C.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l’autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite l’Agenzia Italiana del Farmaco Sito web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
4.9 Sovradosaggio
In caso di sovradosaggio da abemaciclib, possono verificarsi stanchezza e diarrea. Deve essere fornita una terapia di supporto generale.
5.1 Proprietà farmacodinamiche
Categoria farmacoterapeutica: agenti antineoplastici, inibitori delle protein chinasi, codice ATC: L01EF03
Meccanismo d’azione
Abemaciclib è un inibitore potente e selettivo delle chinasi ciclino-dipendenti 4 e 6 (CDK 4 e 6) con maggiore attività nei confronti della Ciclina D1/CDK4 in saggi enzimatici. Abemaciclib previene la fosforilazione della proteina del retinoblastoma (Rb), bloccando la progressione del ciclo cellulare dalla fase G1 alla fase S della divisione cellulare, portando alla soppressione della crescita del tumore. In linee cellulari di carcinoma mammario positivo al recettore dell'estrogeno, un'inibizione sostenuta del target con abemaciclib ha impedito il rebound della fosforilazione del Rb con conseguente senescenza cellulare e apoptosi. In vitro, le linee cellulari tumorali Rb-negative e Rb-impoverite sono generalmente meno sensibili ad abemaciclib. In modelli di xenotrapianto di carcinoma mammario, abemaciclib somministrato quotidianamente senza interruzione, a concentrazioni clinicamente rilevanti, da solo o in associazione con antiestrogeni, ha determinato una riduzione delle dimensioni del tumore.
Effetti farmacodinamici
Nelle pazienti oncologiche, abemaciclib inibisce le CDK4 e CDK6 con inibizione della fosforilazione del Rb e delle topoisomerasi II alfa, che determina l'inibizione del ciclo cellulare a monte del punto di restrizione G1.
Elettrofisiologia cardiaca
L'effetto di abemaciclib sull'intervallo QTcF è stato valutato in 144 pazienti con carcinoma avanzato. Nessun grande cambiamento nell'intervallo QTcF (cioè > 20 ms) è stato rilevato alla massima concentrazione media di abemaciclib osservata allo stato stazionario a seguito dello schema di dosaggio terapeutico. In un'analisi della risposta in base all’esposizione in soggetti sani, a esposizioni paragonabili a una dose di 200 mg due volte al giorno, abemaciclib non ha prolungato l'intervallo QTcF a qualsiasi estensione clinicamente rilevante.
Efficacia clinica e sicurezza
Carcinoma mammario in fase iniziale
Studio randomizzato di Fase 3 monarchE: Verzenios in associazione con terapia endocrina
L'efficacia e la sicurezza di Verzenios in associazione con la terapia endocrina adiuvante sono state valutate nello studio monarchE, uno studio randomizzato, in aperto, a due coorti, di fase 3, in donne e uomini con carcinoma mammario in fase iniziale HR positivo, HER2 negativo, linfonodopositivo ad alto rischio di recidiva. Nella coorte 1, l'alto rischio di recidiva è stato definito da caratteristiche cliniche e patologiche: ≥ 4 pALN (linfonodi ascellari positivi) o 1-3 pALN, e almeno uno dei seguenti criteri: dimensione del tumore ≥ 5 cm o grado istologico 3.
Un totale di 5 637 pazienti è stato randomizzato in un rapporto 1:1 a ricevere per 2 anni Verzenios 150 mg due volte al giorno più una terapia endocrina standard a scelta del medico o la sola terapia endocrina standard. La randomizzazione è stata stratificata in base alla precedente chemioterapia, allo stato della menopausa e alla regione. Gli uomini sono stati stratificati come postmenopausali. I pazienti avevano completato la terapia locoregionale definitiva (con o senza chemioterapia neoadiuvante o adiuvante). I pazienti dovevano essersi ripresi dagli effetti indesiderati acuti di qualsiasi chemioterapia o radioterapia precedente. È stato richiesto un periodo di washout di 21 giorni dopo la chemioterapia e di 14 giorni dopo la radioterapia prima della randomizzazione. Ai pazienti era consentito ricevere fino a 12 settimane di terapia endocrina adiuvante prima della randomizzazione dopo l’ultima terapia non endocrina (intervento chirurgico, chemioterapia o radioterapia). Il trattamento adiuvante con fulvestrant non era consentito come terapia endocrina standard. Erano idonei i pazienti con Performance Status 0 o 1 dell'eastern cooperative oncology group (ECOG). I pazienti con storia di TEV sono stati esclusi dallo studio. Dopo la fine del periodo di trattamento in studio, in entrambi i bracci di trattamento i pazienti hanno continuato a ricevere la terapia endocrina adiuvante per una durata cumulativa di almeno 5 anni e fino a 10 anni, se appropriato dal punto di vista medico. Gli agonisti dell'LHRH sono stati somministrati quando clinicamente indicati a donne in pre e perimenopausa e a uomini.
Tra i 5 637 pazienti randomizzati, 5 120 sono stati arruolati nella coorte 1, che rappresenta il 91 % della popolazione ITT. Nella coorte 1, i dati demografici dei pazienti e le caratteristiche del tumore al basale erano bilanciati tra i bracci di trattamento. L'età mediana dei pazienti arruolati era di circa 51 anni (range, 22 - 89 anni), il 15 % dei pazienti aveva 65 anni o più, il 99 % erano donne, il 71 % erano caucasici, il 24 % erano asiatici e il 5 % altro. Il 43 % dei pazienti era in pre o perimenopausa. La maggior parte dei pazienti ha ricevuto una precedente chemioterapia (36 % neoadiuvante, 62 % adiuvante) e una precedente radioterapia (96 %). La terapia endocrina iniziale ricevuta dai pazienti includeva letrozolo (39 %), tamoxifene (31 %), anastrozolo (22 %) o exemestane (8 %).
Il 65 % dei pazienti aveva 4 o più linfonodi positivi, il 41 % aveva un tumore di Grado 3 e il 24 % aveva dimensioni patologiche del tumore ≥ 5 cm alla chirurgia.
L'endpoint primario era la sopravvivenza libera da malattia invasiva (invasive disease-free survival - IDFS) nella popolazione ITT definita come il tempo dalla randomizzazione alla prima occorrenza di recidiva di tumore mammario invasivo ipsilaterale, di recidiva regionale di carcinoma mammario invasivo, di recidiva a distanza, di carcinoma mammario invasivo controlaterale, occorenza di un secondo tumore primitivo invasivo diverso dal tumore della mammella o morte attribuibile ad una qualsiasi causa. L'endpoint secondario principale era la sopravvivenza libera da recidiva a distanza (distant relapse free survival - DRFS) nella popolazione ITT definita come il tempo dalla randomizzazione al primo avvenimento di recidiva a distanza o morte attribuibile ad una qualsiasi causa.
L'obiettivo primario dello studio è stato raggiunto all'analisi ad interim pianificata (cut-off del 16 marzo 2020). È stato osservato un miglioramento statisticamente significativo dell'IDFS nei pazienti che avevano ricevuto Verzenios più la terapia endocrina rispetto alla sola terapia endocrina nella popolazione ITT. Al momento della successiva analisi finale della sopravvivenza globale (OS, Overall Survival; cut-off del 15 luglio 2025) è stato osservato anche un miglioramento statisticamente significativo dell’OS nei pazienti che avevano ricevuto Verzenios più la terapia endocrina rispetto alla sola terapia endocrina nella popolazione ITT. L'approvazione è stata concessa per la sottopopolazione ampia, la coorte 1.
Nell’analisi finale dell’OS tutti i pazienti nella coorte 1 erano fuori dal periodo di trattamento dello studio di 2 anni e la durata mediana del follow-up era di 76 mesi (6,3 anni).
I risultati di efficacia nella coorte 1 sono riassunti nella Tabella 9, nella Figura 1 e nella Figura 2.
Tabella 9. monarchE: Riassunto dei dati di efficacia (Popolazione della coorte 1)

Abbreviazione: IC = intervallo di confidenza.
Data di cut off dei dati 15 luglio 2025
Figura 1. monarchE: Kaplan-Meier dell'IDFS (Valutazione dello sperimentatore, popolazione
della coorte 1)

Abbreviazioni: IDFS = invasive disease-free survival, sopravvivenza libera da malattia invasiva;
N = numero di pazienti nella popolazione.
Data di cut off dei dati 15 luglio 2025
Figura 2. monarchE: Kaplan-Meier dell’OS (popolazione della coorte 1)

Abbreviazioni: TE = terapia endocrina; OS = sopravvivenza globale; N = numero di pazienti nella popolazione.
Data di cut-off dei dati 15 luglio 2025
Il beneficio è stato osservato in tutti i sottogruppi di pazienti definiti in base alla regione geografica, stato menopausale e precedente chemioterapia all'interno della coorte 1.
Carcinoma mammario avanzato o metastatico
Studio randomizzato di Fase 3 MONARCH 3: Verzenios in associazione con inibitori dell’aromatasi
L'efficacia e la sicurezza di Verzenios in associazione con un inibitore dell’aromatasi (anastrololo o letrozolo) sono state valutate nello studio MONARCH 3, uno studio di fase 3, randomizzato, in doppio cieco, controllato verso placebo, in donne con carcinoma mammario localmente avanzato o metastatico, HR positivo, HER2 negativo che non avevano ricevuto una precedente terapia sistemica per questa patologia. Le pazienti sono state randomizzate in un rapporto 2:1 a ricevere Verzenios 150 mg due volte al giorno più un inibitore dell’aromatasi non steroideo somministrato giornalmente alla dose raccomandata versus placebo più un inibitore non steroideo dell’aromatasi secondo la stessa schedula. L'endpoint primario era la sopravvivenza libera da progressione (PFS) valutata dallo sperimentatore secondo RECIST 1.1; i principali endpoint secondari di efficacia comprendevano il tasso di risposta obiettiva (ORR, Objective Response Rate), il tasso di beneficio clinico (CBR, Clinical
Benefit Rate) e la sopravvivenza globale (OS, Overall Survival).
L'età mediana delle pazienti arruolate era di 63 anni (range 32 - 88). Nel setting (neo)adiuvante, circa il 39 % delle pazienti aveva ricevuto una chemioterapia e il 44 % aveva ricevuto una terapia ormonale. Le pazienti che avevano ricevuto una precedente terapia endocrina (neo)adiuvante dovevano avere completato questa terapia almeno 12 mesi prima della randomizzazione nello studio. La maggior parte delle pazienti (96 %) aveva una malattia metastatica al basale. Approssimativamente il 22 % delle pazienti presentava una malattia esclusivamente scheletrica e il 53 % delle pazienti aveva metastasi viscerali.
Lo studio ha raggiunto l'endpoint primario di miglioramento della PFS. I risultati di efficacia primaria sono riassunti nella Tabella 10 e nella Figura 3.
Tabella 10. MONARCH 3: Riassunto dei dati di efficacia (Valutazione dello sperimentatore,
popolazione intent-to-treat)


a Malattia misurabile definite secondo RECIST versione 1.1
b Risposta completa + risposta parziale c Risposta completa + risposta parziale + malattia stabile per > 6 mesi
N=numero di pazienti; IC=intervallo di confidenza; NR=non raggiunto.
Figura 3. MONARCH 3: Kaplan-Meier della sopravvivenza libera da progressione (Valutazione dello sperimentatore, popolazione intent-to-treat)

Questi risultati corrispondono ad una riduzione clinicamente significativa del rischio di progressione della malattia o morte del 46 % per le pazienti trattate con abemaciclib più un inibitore dell'aromatasi.
Il dato di OS non era maturo all'analisi finale della PFS (93 eventi osservati nei due bracci). L'HR era 1,057 (IC 95 %: 0,683, 1,633), p = 0,8017.
Una serie di analisi di sottogruppo predefinite della PFS ha mostrato risultati coerenti fra i sottogruppi di pazienti, compresi l'età (< 65 o ≥ 65 anni), la localizzazione della malattia, il setting della malattia (metastatica de novo vs recidiva metastatica vs recidiva localmente avanzata), la presenza di malattia misurabile, lo stato del recettore del progesterone e il performace status al basale secondo ECOG. È stata osservata una riduzione del rischio di progressione della malattia o decesso in pazienti con malattia viscerale, (HR di 0,567 [IC 95 %: 0,407, 0,789]), PFS mediana 21,6 mesi rispetto a 14,0 mesi, in pazienti con malattia esclusivamente scheletrica (HR di 0,565, [IC 95 %: 0,306, 1,044]) e in pazienti con malattia misurabile (HR di 0,517, [IC 95 %: 0,392, 0,681]).
Alla prima analisi ad interim dell’OS, sono stati osservati 197 eventi tra i due bracci e l'HR era 0,786 (IC 95 %: 0,589, 1,049).
Alla seconda analisi ad interim dell’OS, sono stati osservati 255 eventi tra i due bracci e l'HR era 0,754 (IC 95 %: 0,584, 0,974).
I risultati dell'analisi finale della OS non hanno raggiunto la significatività statistica (riassunti nella Tabella 11 e nella Figura 4).
Tabella 11.: MONARCH 3: Riassunto dei dati di sopravvivenza globale (popolazione intent-to-treat)

Abbreviazioni: N = numero di pazienti; IC = intervallo di confidenza; ITT = intent-to-treat; OS (overall survival) = sopravvivenza globale
Le analisi per l’OS nei pazienti con malattia viscerale hanno mostrato un OS HR di 0,758 (IC 95%: 0,558, 1,030). La OS mediana è stata di 63,72 mesi nel braccio abemaciclib più IA e di 48,82 mesi nel braccio placebo più IA. Analogamente alla popolazione ITT, i risultati non erano statisticamente significativi.
Figura 4. MONARCH 3: Kaplan-Meier della sopravvivenza globale (popolazione intent-to-treat)

Studio randomizzato di Fase 3 MONARCH 2: Verzenios in associazione con fulvestrant
L'efficacia e la sicurezza di Verzenios in associazione con fulvestrant sono state valutate nello studio MONARCH 2, studio di fase 3, randomizzato, in doppio cieco, controllato verso placebo in donne con carcinoma mammario localmente avanzato o metastatico, HR positivo, HER2 negativo. Le pazienti sono state randomizzate in un rapporto 2:1 a ricevere Verzenios 150 mg due volte al giorno più fulvestrant 500 mg ad intervalli di un mese, con una dose aggiuntiva di 500 mg somministrata due settimane dopo la dose iniziale, rispetto a placebo più fulvestrant secondo la stessa schedula.
L'endpoint primario era la PFS valutata dallo sperimentatore secondo RECIST 1.1; i principali
endpoint secondari di efficacia comprendevano l’ORR, il CBR e l’OS.
L'età mediana delle pazienti arruolate era di 60 anni (range, 32 - 91 anni). In ciascun braccio di trattamento la maggior parte delle pazienti era di razza bianca e nessuna aveva ricevuto una chemioterapia per la malattia metastatica. Il 17 % delle pazienti era in fase pre/perimenopausale con soppressione ovarica indotta con un agonista del GnRH. Circa il 56 % delle pazienti aveva metastasi viscerali. Circa il 25 % delle pazienti presentava una resistenza endocrina primaria (progressione in corso di terapia endocrina entro i primi 2 anni di terapia endocrina adiuvante o entro i primi 6 mesi di terapia endocrina di prima linea per carcinoma mammario metastatico) mentre nella maggior parte, la resistenza endocrina si è sviluppata in seguito. Il 59 % delle pazienti ha ricevuto la terapia endocrina più recente nel setting (neo)adiuvante e il 38 % in quello metastatico.
Lo studio ha raggiunto l'endpoint primario di miglioramento della PFS. I risultati di efficacia primaria sono riassunti nella Tabella 12 e nella Figura 5.
Tabella 12. MONARCH 2: Riassunto dei dati di efficacia (Valutazione dello sperimentatore,
popolazione intent-to-treat)

a Malattia misurabile definita secondo RECIST versione 1.1
b Risposta completa + risposta parziale
c Risposta completa + risposta parziale + malattia stabile per > 6 mesi
N = numero di pazienti; IC = intervallo di confidenza; NR = non raggiunto
Figura 5. MONARCH 2: Kaplan-Meier della sopravvivenza libera da progressione (Valutazione dello sperimentatore, popolazione intent-to-treat)

Questi risultati corrispondono ad una riduzione clinicamente significativa del rischio di progressione della malattia o morte del 44,7 % per le pazienti trattate con Verzenios più fulvestrant. Verzenios più fulvestrant ha prolungato la sopravvivenza libera da progressione senza un peggioramento clinicamente significativo o importante della qualità di vita correlata allo stato di salute.
Una serie di analisi di sottogruppo predefinite della PFS ha mostrato risultati coerenti fra i sottogruppi di pazienti, compresi l’età (< 65 o ≥ 65 anni), la razza, la regione geografica, la localizzazione della malattia, la resistenza alla terapia endocrina, la presenza di malattia misurabile, lo stato del recettore del progesterone e lo stato menopausale. È stata osservata una riduzione del rischio di progressione della malattia o decesso in pazienti con malattia viscerale (HR 0,481 (IC 95 %: 0,369, 0,627), PFS mediana di 14,7 mesi verso 6,5 mesi), in pazienti con sola malattia ossea (HR di 0,543 (IC 95 %: 0,355, 0,833), in pazienti con malattia misurabile (HR 0,523 [IC 95 %: 0,412, 0,644]). Nelle pazienti che erano in fase pre/perimenopausale, l’hazard ratio era 0,415 (IC 95 %: 0,246, 0,698), nelle pazienti con recettore del progesterone negativo, HR era di 0,509 (IC 95 %: 0,325, 0,797).
La PFS si è dimostrata coerente anche nella sotto-popolazione con malattia localmente avanzata o metastatica che non aveva ricevuto una precedente terapia endocrina.
All'analisi ad interim predefinita dell’OS (cut-off del 20 giugno 2019) la popolazione ITT ha mostrato un miglioramento statisticamente significativo nei pazienti trattati con Verzenios più fulvestrant rispetto a quelli che hanno ricevuto placebo più fulvestrant. I risultati relativi all’OS sono riassunti nella Tabella 13.
Tabella 13. MONARCH 2: Riassunto dei dati sulla sopravvivenza globale (popolazione Intent-to-treat)

N = numero di pazienti; IC = intervallo di confidenza; OS (overall survival) = sopravvivenza globale
Le analisi per l’OS in base ai fattori di stratificazione hanno mostrato un HR per l’OS di
0,675 (IC 95 %: 0,511, 0,891) in pazienti con malattia viscerale e 0,686 (IC 95 %: 0,451, 1,043) in pazienti con resistenza endocrina primaria.
All'analisi finale predefinita della OS (cut-off del 18 marzo 2022), sono stati osservati 440 eventi nei 2 bracci. Il miglioramento della OS precedentemente osservato all'analisi ad interim della OS (cut-off del 20 giugno 2019) è stato mantenuto nel braccio abemaciclib più fulvestrant rispetto al braccio placebo più fulvestrant, con un HR di 0,784 (IC 95 %: 0,644, 0,955). La OS mediana è stata di 45,8 mesi nel braccio abemaciclib più fulvestrant e di 37,25 mesi nel braccio placebo più fulvestrant. I risultati relativi all’OS sono presentati nella Figura 6.
Figura 6. MONARCH 2: Kaplan-Meier della sopravvivenza globale (popolazione Intent-to-treat)

Popolazione pediatrica
L'Agenzia europea per i medicinali ha previsto l'esonero dall'obbligo di presentare i risultati degli studi con Verzenios in tutti i sottogruppi della popolazione pediatrica nel cancro al seno (vedere paragrafo 4.2 per informazioni sull'uso pediatrico).
L'efficacia e la sicurezza di Verzenios in associazione con irinotecan e temozolomide sono state valutate nello studio J1S-MC-JP04, uno studio multicentrico, randomizzato, in aperto, di fase 2 in partecipanti con sarcoma di Ewing recidivante o refrattario. L'endpoint primario era la sopravvivenza libera da progressione (PFS) valutata da un comitato di revisione indipendente in cieco. 46 partecipanti, di età compresa tra 3 e 35 anni, sono stati randomizzati a ricevere Verzenios più irinotecan e temozolomide o irinotecan e temozolomide in un rapporto 2:1. Il 58,7 % dei pazienti (27 pazienti) aveva < 18 anni. 45 partecipanti sono stati trattati in cicli di 21 giorni fino alla progressione della malattia o al verificarsi di altri criteri di interruzione. Non è stata osservata alcuna differenza nella PFS con l'aggiunta di Verzenios. La PFS mediana è stata di 2,8 mesi nei pazienti trattati con Verzenios in associazione con irinotecan e temozolomide e di 2,9 mesi nei pazienti trattati con irinotecan e temozolomide (HR 0,64 [IC 95 %: 0,28; 1,45]).
5.2 Proprietà farmacocinetiche
Assorbimento
L'assorbimento di abemaciclib è lento, con un Tmax di 8 ore e una biodisponibilità assoluta media di circa il 45 %. Nell'intervallo di dosaggio terapeutico di 50-200 mg, l'aumento dell'esposizione plasmatica (AUC) e della Cmax è approssimativamente proporzionale alla dose. Lo stato stazionario è stato raggiunto entro 5 giorni dopo la somministrazione ripetuta due volte al giorno e abemaciclib si è accumulato e i valori della media geometrica del fattore di accumulo erano 3,7 (58 % CV) e 5,8 (65 % CV), rispettivamente per la Cmax e l’AUC. Un pasto ricco di grassi ha aumentato l'AUC di abemaciclib e dei suoi metaboliti attivi del 9 % ed ha aumentato la Cmax del 26 %. Questi cambiamenti non sono stati considerati clinicamente rilevanti. Pertanto, abemaciclib può essere assunto con o senza cibo.
Distribuzione
Abemaciclib è altamente legato alle proteine plasmatiche negli esseri umani (frazione legata media approssimativamente dal 96 % al 98 %). La media geometrica del volume di distribuzione sistemico è di circa 750 L (69 % CV), che indica la distribuzione di abemaciclib nei tessuti. Le concentrazioni di abemaciclib e dei suoi metaboliti attivi nel liquido cerebrospinale sono paragonabili alle concentrazioni plasmatiche libere.
Biotrasformazione
Il metabolismo epatico è la principale via di clearance di abemaciclib. Abemaciclib è metabolizzato in diversi metaboliti principalmente dal citocromo P450 (CYP) 3A4. La biotrasformazione primaria è l’idrossilazione ad un metabolita che rimane in circolo con una AUC pari al 77 % di quella del farmaco originario. Inoltre, i metaboliti N-desetil e N-desetil-idrossi rimangono in circolo con una AUC pari al 39 % e al 15 % del farmaco originario. Questi metaboliti circolanti sono attivi con una potenza simile a quella di abemaciclib.
Eliminazione
La clearance epatica media geometrica (CL) di abemaciclib era 21,8 L/h (39,8 % CV) e l'emivita
media di eliminazione plasmatica di abemaciclib nei pazienti era di 24,8 ore (52,1 % CV). Dopo una singola dose orale di [14C] -abemaciclib, circa l'81 % della dose è stata escreta nelle feci e il 3,4 % è stata escreta nelle urine. La maggior parte della dose eliminata nelle feci era costituita da metaboliti.
Popolazioni speciali
Età, genere e peso corporeo
L'età, il genere e il peso corporeo non hanno avuto alcun effetto sull'esposizione di abemaciclib in un'analisi farmacocinetica di popolazione in pazienti oncologici (135 maschi e 859 femmine, fascia d'età 24-91 anni e peso corporeo 36 - 175 kg).
Compromissione epatica
Abemaciclib è metabolizzato nel fegato. Una compromissione epatica lieve (Child Pugh A) e
moderata (Child Pugh B) non hanno avuto alcun effetto sull'esposizione di abemaciclib. Nei soggetti con compromissione epatica severa (Child Pugh C), l'AUC0-∞ di abemaciclib e la frazione libera di abemaciclib e dei suoi metaboliti attivi, corretta per la potenza, sono aumentati, rispettivamente, di 2,1 volte e di 2,4 volte. L'emivita di abemaciclib è aumentata da 24 a 55 ore (vedere paragrafo 4.2).
Compromissione renale
La clearance renale di abemaciclib e dei suoi metaboliti è minore. Una compromissione renale lieve e moderata non hanno avuto alcun effetto sull'esposizione di abemaciclib. Non ci sono dati in pazienti con compromissione renale severa, malattia renale allo stadio terminale o in pazienti in dialisi.
5.3 Dati preclinici di sicurezza
Gli effetti principali a carico degli organi bersaglio di potenziale rilevanza per gli esseri umani si sono verificati nel tratto gastrointestinale, negli organi emolinfopoietici e nel tratto riproduttivo maschile in topi, ratti e cani in studi con durata fino a 13 settimane. Gli effetti sugli occhi e sulle valvole cardiache si sono verificati solo nei roditori a livelli di esposizione clinicamente rilevanti. Effetti sul polmone e sulla muscolatura scheletrica si sono verificati solo nei roditori a livelli di esposizione almeno 2 volte superiori ai livelli di esposizione umana. Effetti sul rene si sono verificati solo nei roditori a livelli di esposizione almeno 6 volte superiori ai livelli di esposizione umana. È stato osservato il recupero completo o parziale per tutti gli effetti sugli organi bersaglio alla fine del periodo di recupero di 28 giorni, ad eccezione degli effetti sul tratto riproduttivo maschile.
Genotossicità
Abemaciclib non è risultato mutageno in un test di reversione di una mutazione batterica (Ames), non è risultato clastogenico in un test di aberrazione cromosomica in vitro su linfociti ematici periferici umani e non è risultato clastogenico in un test del micronucleo del midollo osseo di ratto in vivo.
Carcinogenicità
Abemaciclib è stato valutato per la carcinogenicità in studi della durata di 2 anni su ratti e topi. Nei ratti maschi, la somministrazione orale quotidiana di abemaciclib ha provocato adenomi benigni delle cellule interstiziali testicolari a esposizioni di circa 1,5 volte l'esposizione clinica umana. Inoltre, è stata osservata iperplasia delle cellule interstiziali a esposizioni di circa 0,1 volte l'esposizione clinica umana. Non è noto se questi effetti si verificheranno nell’uomo. Non sono stati rilevati reperti neoplastici nei topi o nelle femmine di ratto dovuti alla somministrazione di abemaciclib.
Compromissione della fertilità
Abemaciclib può compromettere la fertilità nei maschi in età fertile. In studi di tossicità a dosi ripetute fino a 3 mesi di durata, i risultati correlati ad abemaciclib nel testicolo, nell'epididimo, nella prostata e nelle vescicole seminali includevano diminuzione del peso degli organi, detriti cellulari intratubulari, ipospermia, dilatazione tubulare, atrofia e egenerazione/necrosi. Questi effetti si sono verificati nei ratti e nei cani a esposizioni rispettivamente di circa 2 e 0,02 volte l'esposizione clinica nell'uomo. In uno studio sulla fertilità maschile nel ratto, abemaciclib non ha avuto effetti sulle prestazioni riproduttive.
In uno studio sulla fertilità femminile nel ratto e sullo sviluppo embrionale precoce e in studi di tossicità a dosi ripetute, abemaciclib non ha avuto alcun effetto sulla prestazione riproduttiva né alcun effetto importante sul tratto riproduttivo femminile indicativo di un rischio di ridotta fertilità nelle femmine.
Tossicità dello sviluppo
Abemaciclib è risultato teratogeno e ha causato una riduzione del peso fetale ad un’esposizione materna simile alla dose raccomandata nell'uomo.
6.1 Elenco degli eccipienti
Nucleo della compressa
Sodio croscarmelloso
Lattosio monoidrato
Cellulosa microcristallina
Silice colloidale idrata
Sodio stearil fumarato
Film di rivestimento
Verzenios 50 mg compresse rivestite con film
Alcool polivinilico (E1203)
Titanio diossido (E171)
Macrogol (E1521)
Talco (E553b)
Ossido di ferro giallo (E172)
Ossido di ferro rosso (E172)
Verzenios 100 mg compresse rivestite con film
Alcool polivinilico (E1203)
Titanio diossido (E171)
Macrogol (E1521)
Talco (E553b)
Verzenios 150 mg compresse rivestite con film
Alcool polivinilico (E1203)
Titanio diossido (E171)
Macrogol (E1521)
Talco (E553b)
Ossido di ferro giallo (E172)
6.2 Incompatibilità
Non applicabile.
6.3 Periodo di validità
3 anni
6.4 Precauzioni particolari per la conservazione
Questo medicinale non richiede alcuna condizione speciale di conservazione.
6.5 Natura e contenuto del contenitore
Blister di PCTFE/PE/PVC sigillati con un foglio di alluminio in confezioni da 14, 28, 42, 56, 70 o
168 compresse rivestite con film.
Blister divisibile per dose unitaria in alluminio/alluminio da 28 x 1 compresse rivestite con film.
È possibile che non tutte le confezioni siano commercializzate.
6.6 Precauzioni particolari per lo smaltimento
Il medicinale non utilizzato e i rifiuti derivati da tale medicinale devono essere smaltiti in conformità alla normativa locale vigente.
Eli Lilly Nederland B.V., Orteliuslaan 1000, 3528 BD Utrecht, Paesi Bassi.
EU/1/18/1307/001
EU/1/18/1307/002
EU/1/18/1307/003
EU/1/18/1307/004
EU/1/18/1307/005
EU/1/18/1307/006
EU/1/18/1307/007
EU/1/18/1307/008
EU/1/18/1307/009
EU/1/18/1307/010
EU/1/18/1307/011
EU/1/18/1307/012
EU/1/18/1307/013
EU/1/18/1307/014
EU/1/18/1307/015
EU/1/18/1307/016
EU/1/18/1307/017
EU/1/18/1307/018
EU/1/18/1307/019
EU/1/18/1307/020
EU/1/18/1307/021
Data della prima autorizzazione: 27 settembre 2018
Data del rinnovo più recente: 23 giugno 2023
29 gennaio 2026
Informazioni più dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia europea per i medicinali, https://www.ema.europa.eu.